

Amino-acid selective isotope labeling enables simultaneous overlapping signal decomposition and information extraction from NMR spectra
- PMID: 32002710
- PMCID: PMC7080692
- DOI: 10.1007/s10858-019-00295-9
Amino-acid selective isotope labeling enables simultaneous overlapping signal decomposition and information extraction from NMR spectra
Abstract
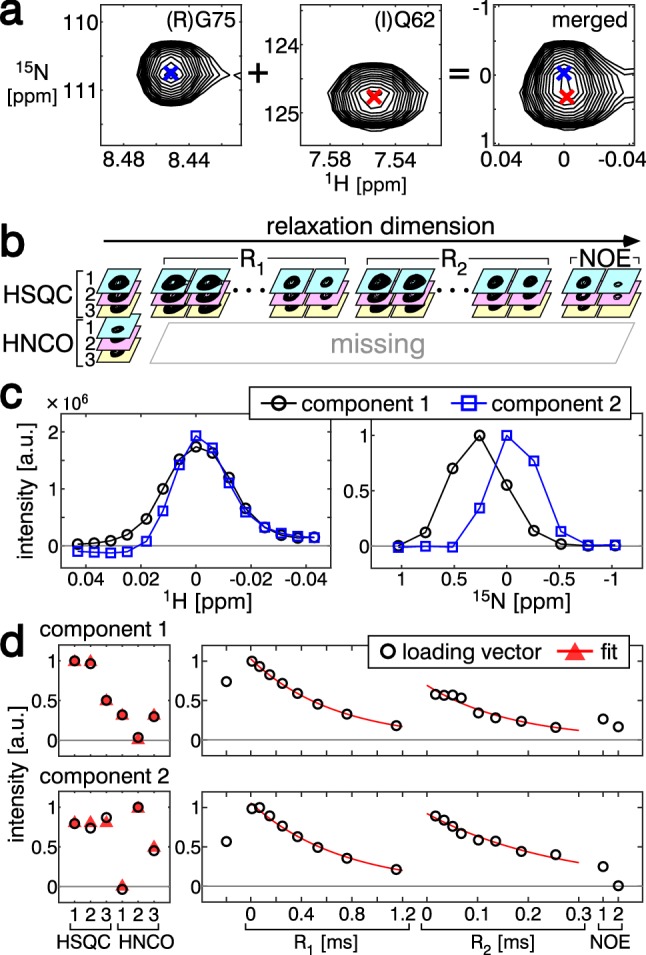
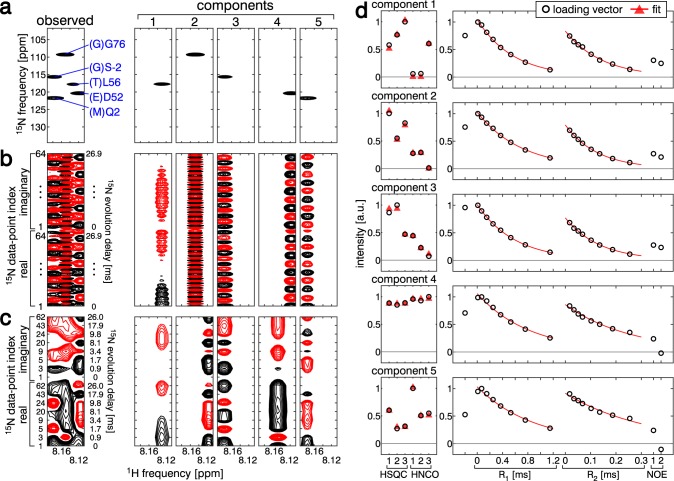
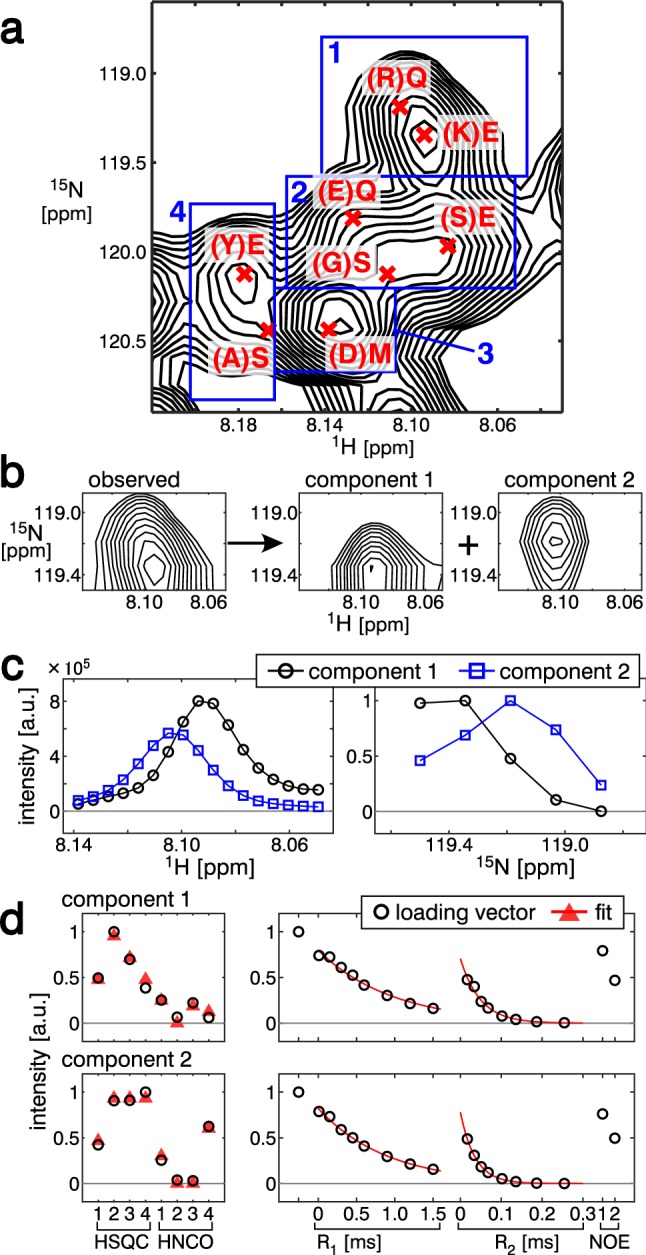
Signal overlapping is a major bottleneck for protein NMR analysis. We propose a new method, stable-isotope-assisted parameter extraction (SiPex), to resolve overlapping signals by a combination of amino-acid selective isotope labeling (AASIL) and tensor decomposition. The basic idea of Sipex is that overlapping signals can be decomposed with the help of intensity patterns derived from quantitative fractional AASIL, which also provides amino-acid information. In SiPex, spectra for protein characterization, such as 15N relaxation measurements, are assembled with those for amino-acid information to form a four-order tensor, where the intensity patterns from AASIL contribute to high decomposition performance even if the signals share similar chemical shift values or characterization profiles, such as relaxation curves. The loading vectors of each decomposed component, corresponding to an amide group, represent both the amino-acid and relaxation information. This information link provides an alternative protein analysis method that does not require "assignments" in a general sense; i.e., chemical shift determinations, since the amino-acid information for some of the residues allows unambiguous assignment according to the dual selective labeling. SiPex can also decompose signals in time-domain raw data without Fourier transform, even in non-uniformly sampled data without spectral reconstruction. These features of SiPex should expand biological NMR applications by overcoming their overlapping and assignment problems.
Keywords: Combinatorial selective labeling; Non-uniform sampling (NUS); Relaxation analysis; Spectral deconvolution; Stable isotope encoding (SiCode); Tensor factorization.
Conflict of interest statement
Takuma Kasai and Takanori Kigawa are inventors on a Japanese Patent (Number 6191927) related to this work. Shunsuke Ono, Seizo Koshiba, Masayuki Yamamoto, Toshiyuki Tanaka, and Shiro Ikeda declare that they have no conflict of interest.
Figures

References
-
- Andersson CA, Bro R. The N-way toolbox for MATLAB. Chemometrics Intellig Lab Syst. 2000;52:1–4. doi: 10.1016/S0169-7439(00)00071-X. - DOI
-
- Barna JCJ, Laue ED. Conventional and exponential sampling for 2D NMR experiments with application to a 2D NMR spectrum of a protein. J Magn Reson. 1969;75:384–389.
-
- Barna JCJ, Laue ED, Mayger MR, Skilling J, Worrall SJP. Exponential sampling, an alternative method for sampling in two-dimensional NMR experiments. J Magn Reson. 1987;73:69–77.
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
