

Evaluation of X-Linked Adrenoleukodystrophy Newborn Screening in North Carolina
- PMID: 32003821
- PMCID: PMC7042889
- DOI: 10.1001/jamanetworkopen.2019.20356
Evaluation of X-Linked Adrenoleukodystrophy Newborn Screening in North Carolina
Abstract
Importance: X-linked adrenoleukodystrophy (X-ALD) is a peroxisomal genetic disorder in which an accumulation of very long-chain fatty acids leads to inflammatory demyelination in the central nervous system and to adrenal cortex atrophy. In 2016, X-ALD was added to the US Recommended Uniform Screening Panel.
Objective: To evaluate the performance of a single-tier newborn screening assay for X-ALD in North Carolina.
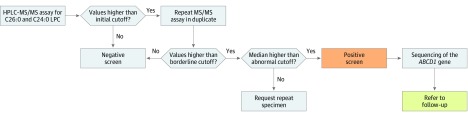
Design, setting, and participants: This diagnostic screening study was of all newborn dried blood spot specimens received in the North Carolina State Laboratory of Public Health between January 2 and June 1, 2018, excluding specimens of insufficient quantity or quality. A total of 52 301 specimens were screened for X-ALD using negative ionization high-performance liquid chromatography tandem mass spectrometry to measure C24:0- and C26:0-lysophosphatidylcholine concentrations. Sanger sequencing of the adenosine triphosphate-binding cassette subfamily D member 1 (ABCD1) gene was performed on screen-positive specimens.
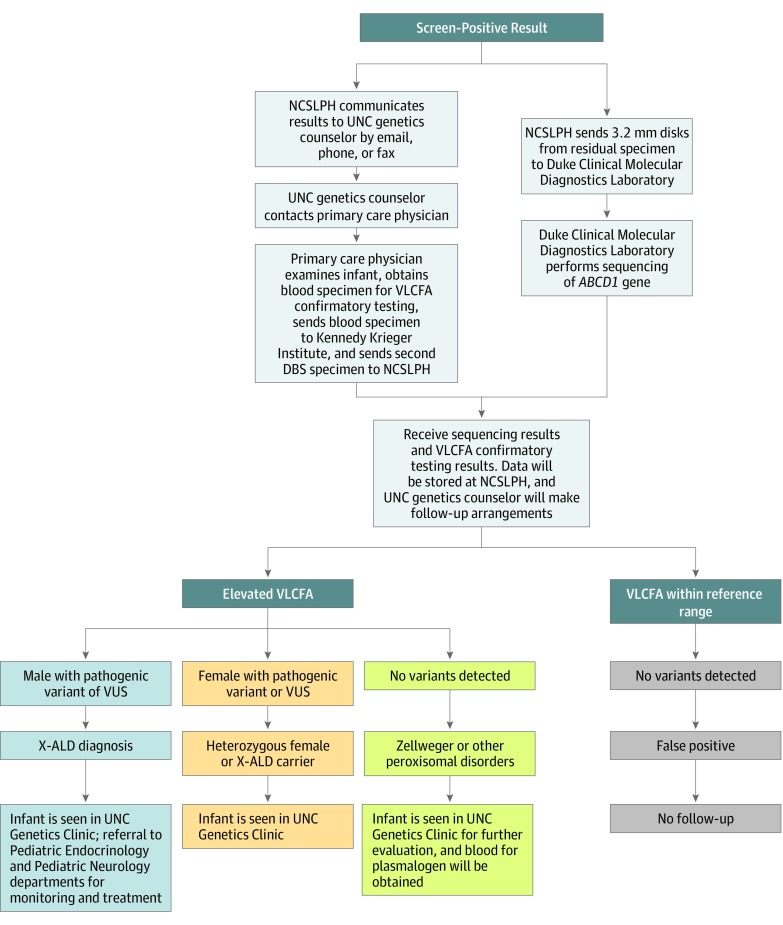
Exposures: A medical and family history, newborn physical examination, sequencing of ABCD1 on dried blood spot samples, and plasma analysis of very long-chain fatty acids were obtained for all infants with screen-positive results.
Main outcomes and measures: The prevalence of X-ALD in North Carolina and the positive predictive value and false-positive rate for the first-tier assay were determined.
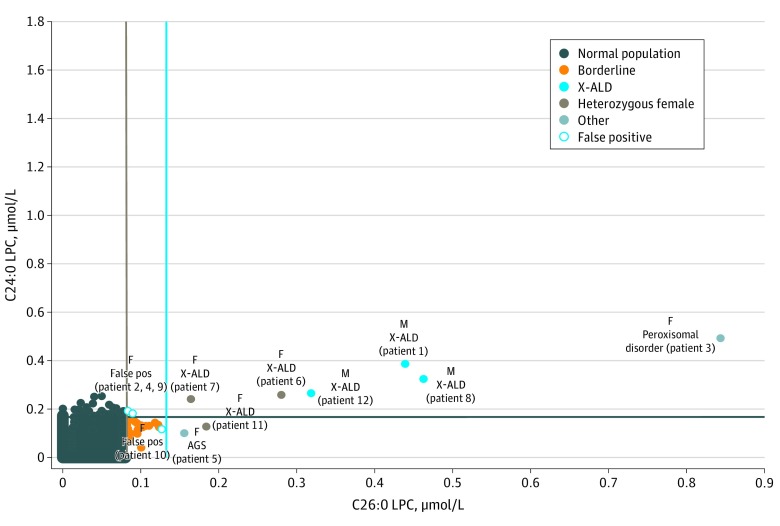
Results: Of 52 301 infants tested (47.8% female, 50.6% male, and 1.7% other or unknown sex), 12 received screen-positive results. Of these 12 infants, 8 were confirmed with a genetic disorder: 3 male infants with X-ALD, 3 X-ALD-heterozygous female infants, 1 female infant with a peroxisome biogenesis disorder, and 1 female infant with Aicardi-Goutières syndrome. Four infants were initially classified as having false-positives results, including 3 female infants who were deemed unaffected and 1 male infant with indeterminate results on confirmatory testing. The positive predictive value for X-ALD or other genetic disorders for the first-tier assay was 67%, with a false-positive rate of 0.0057%.
Conclusions and relevance: This newborn screening pilot study reported results on 2 lysophosphatidylcholine analytes, identifying 3 male infants with X-ALD, 3 X-ALD-heterozygous female infants, and 3 infants with other disorders associated with increased very long-chain fatty acids. These results showed successful implementation in a public health program with minimal risk to the population. The findings will support other state laboratories planning to implement newborn screening for X-ALD and related disorders.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
