

Chromatin Organization in Early Land Plants Reveals an Ancestral Association between H3K27me3, Transposons, and Constitutive Heterochromatin
- PMID: 32004456
- PMCID: PMC7209395
- DOI: 10.1016/j.cub.2019.12.015
Chromatin Organization in Early Land Plants Reveals an Ancestral Association between H3K27me3, Transposons, and Constitutive Heterochromatin
Abstract
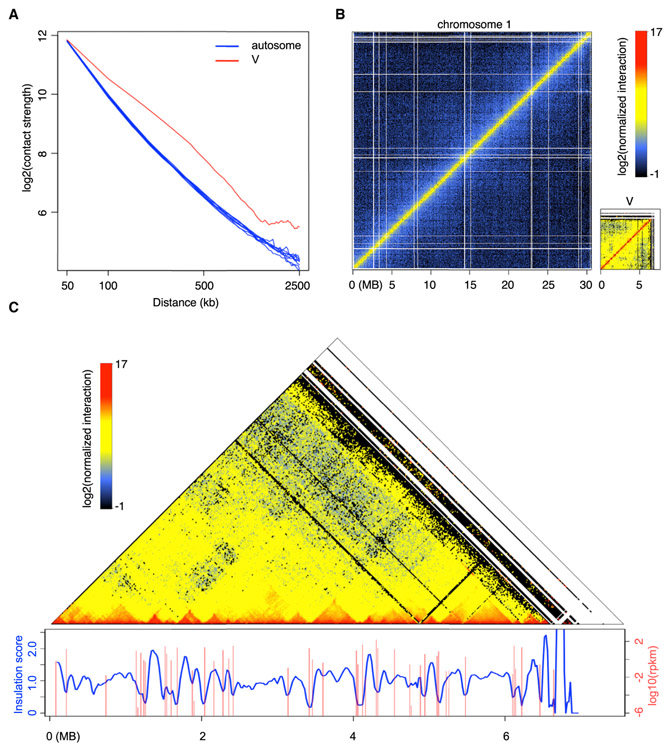
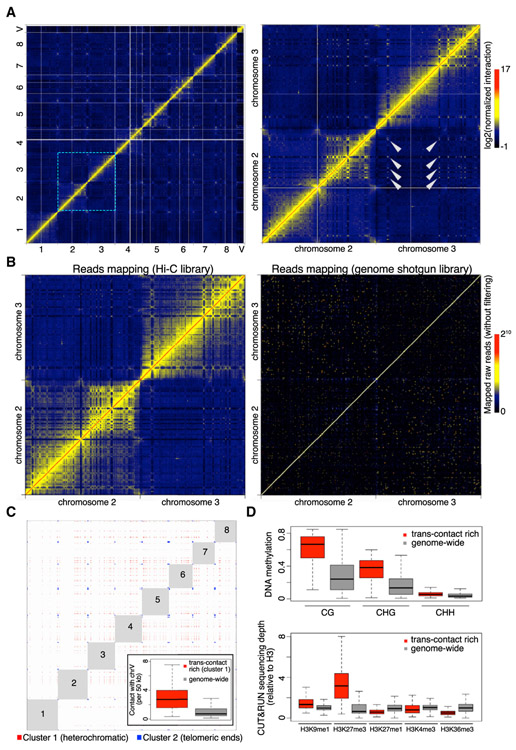
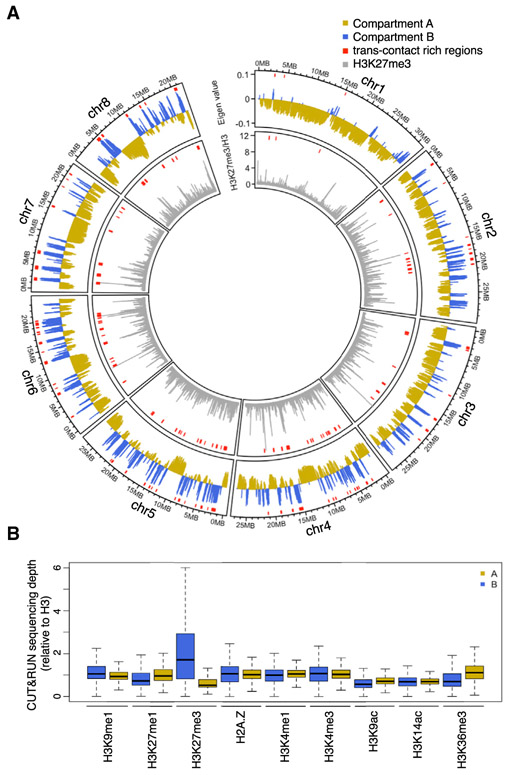
Genome packaging by nucleosomes is a hallmark of eukaryotes. Histones and the pathways that deposit, remove, and read histone modifications are deeply conserved. Yet, we lack information regarding chromatin landscapes in extant representatives of ancestors of the main groups of eukaryotes, and our knowledge of the evolution of chromatin-related processes is limited. We used the bryophyte Marchantia polymorpha, which diverged from vascular plants circa 400 mya, to obtain a whole chromosome genome assembly and explore the chromatin landscape and three-dimensional genome organization in an early diverging land plant lineage. Based on genomic profiles of ten chromatin marks, we conclude that the relationship between active marks and gene expression is conserved across land plants. In contrast, we observed distinctive features of transposons and other repetitive sequences in Marchantia compared with flowering plants. Silenced transposons and repeats did not accumulate around centromeres. Although a large fraction of constitutive heterochromatin was marked by H3K9 methylation as in flowering plants, a significant proportion of transposons were marked by H3K27me3, which is otherwise dedicated to the transcriptional repression of protein-coding genes in flowering plants. Chromatin compartmentalization analyses of Hi-C data revealed that repressed B compartments were densely decorated with H3K27me3 but not H3K9 or DNA methylation as reported in flowering plants. We conclude that, in early plants, H3K27me3 played an essential role in heterochromatin function, suggesting an ancestral role of this mark in transposon silencing.
Keywords: H3K27me3; Marchantia; bryophytes; chromatin; evolution; genome; sex chromosome; transposon.
Crown Copyright © 2019. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests The authors declare no competing interests.
Figures
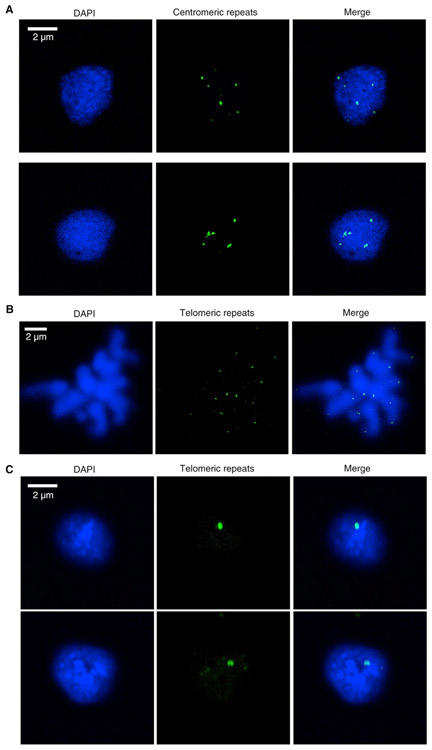
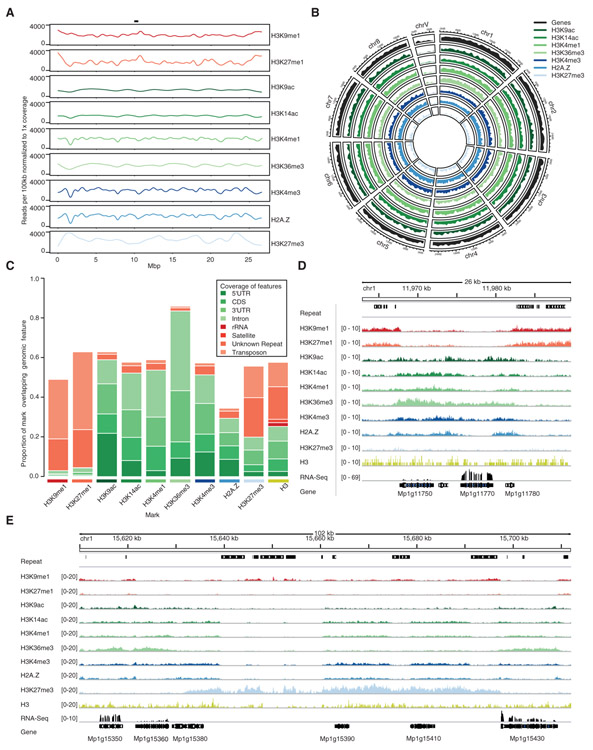
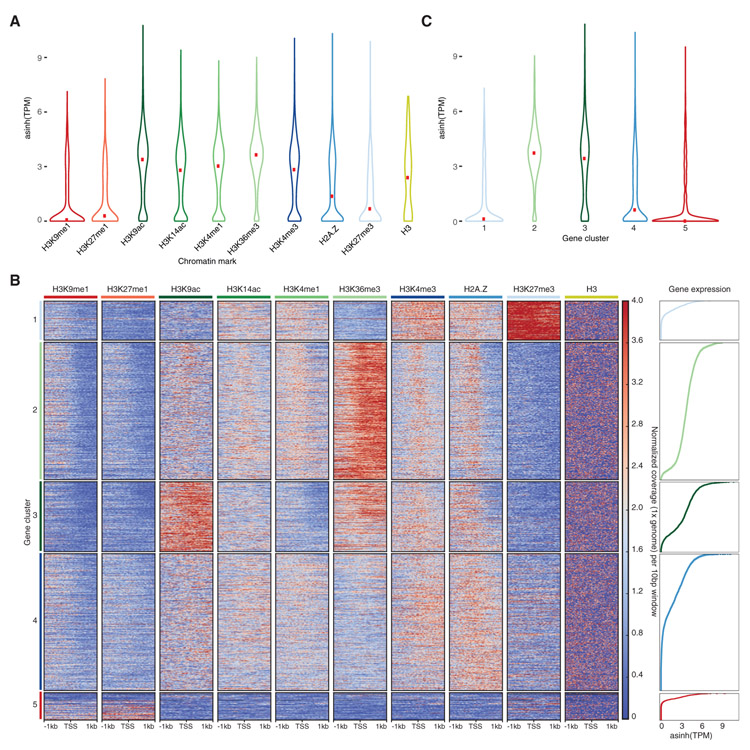
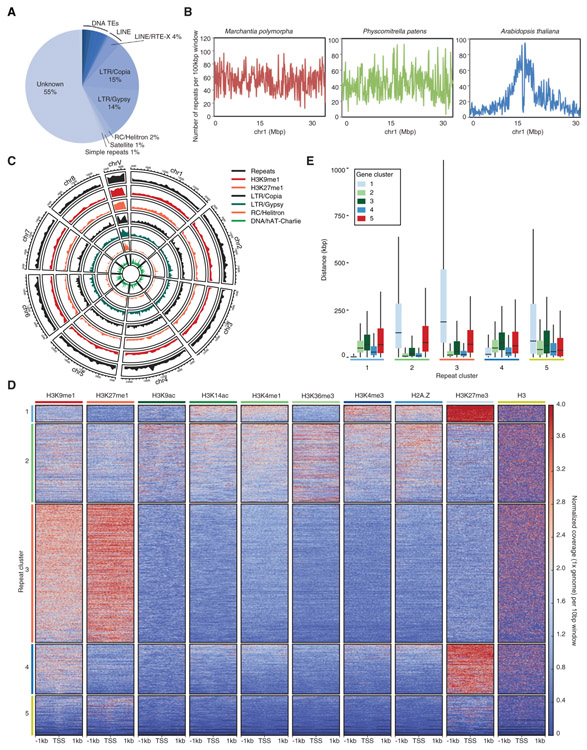
Comment in
-
Evolution: Heterochromatin Diversity in Early-Branching Land Plants.Curr Biol. 2020 Feb 24;30(4):R161-R163. doi: 10.1016/j.cub.2019.12.044. Curr Biol. 2020. PMID: 32097641
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
