

Host-Specific Evolutionary and Transmission Dynamics Shape the Functional Diversification of Staphylococcus epidermidis in Human Skin
- PMID: 32004459
- PMCID: PMC7192218
- DOI: 10.1016/j.cell.2020.01.006
Host-Specific Evolutionary and Transmission Dynamics Shape the Functional Diversification of Staphylococcus epidermidis in Human Skin
Abstract
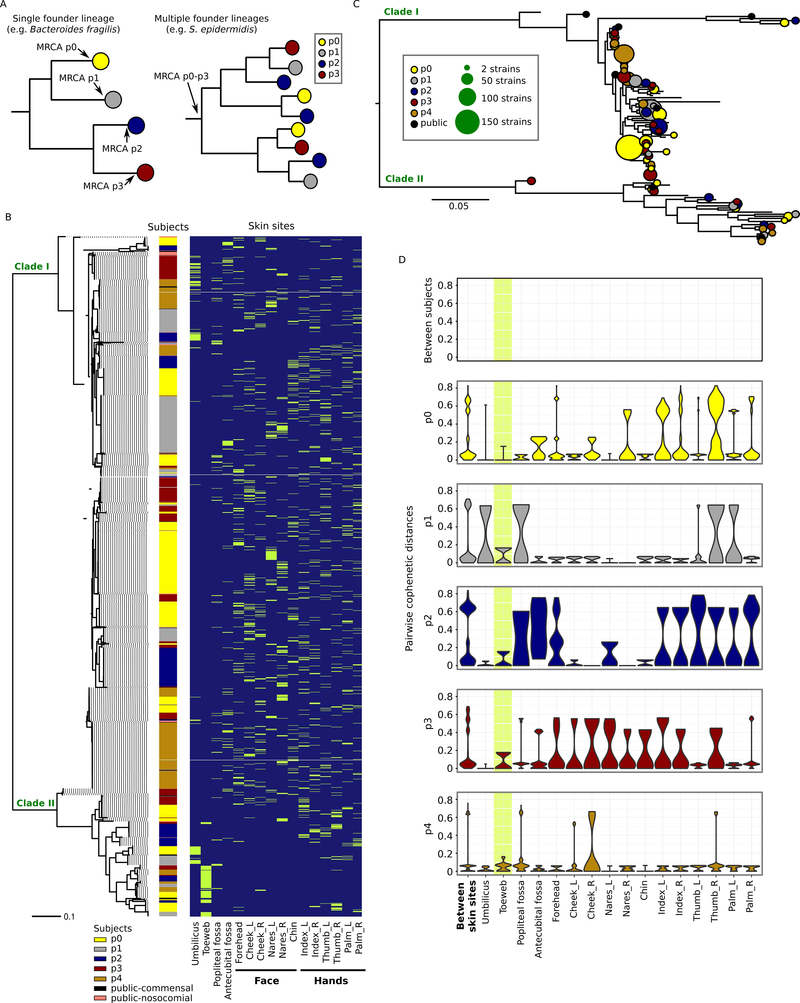
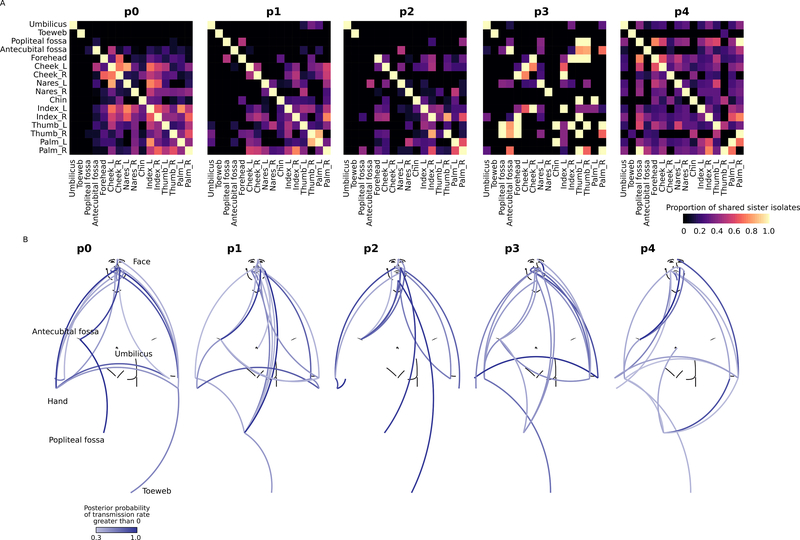
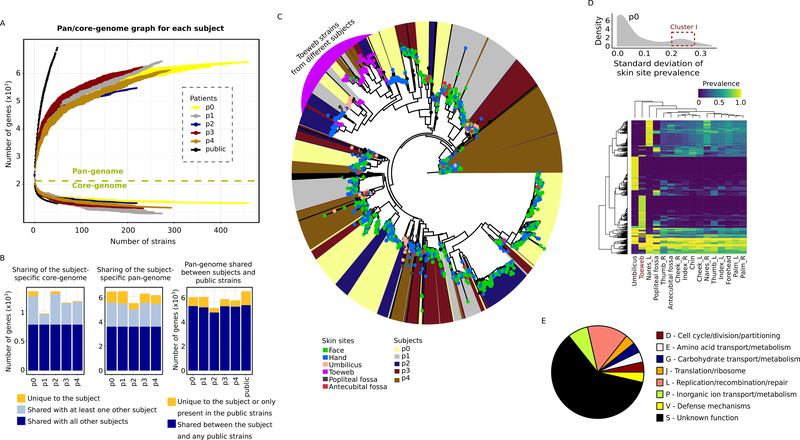
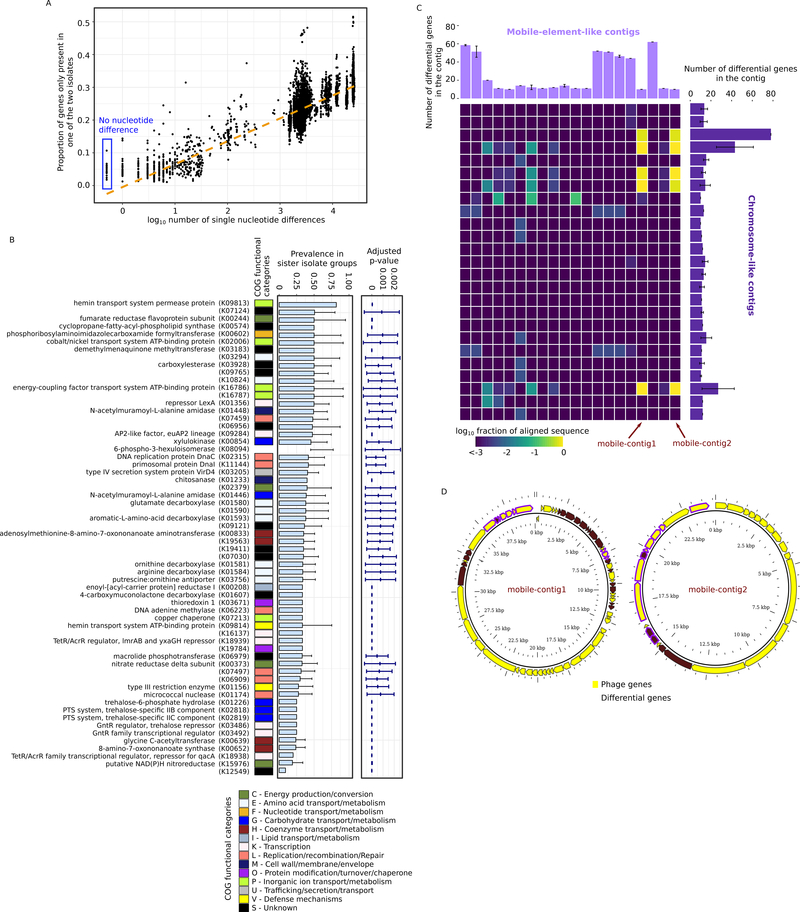
Metagenomic inferences of bacterial strain diversity and infectious disease transmission studies largely assume a dominant, within-individual haplotype. We hypothesize that within-individual bacterial population diversity is critical for homeostasis of a healthy microbiome and infection risk. We characterized the evolutionary trajectory and functional distribution of Staphylococcus epidermidis-a keystone skin microbe and opportunistic pathogen. Analyzing 1,482 S. epidermidis genomes from 5 healthy individuals, we found that skin S. epidermidis isolates coalesce into multiple founder lineages rather than a single colonizer. Transmission events, natural selection, and pervasive horizontal gene transfer result in population admixture within skin sites and dissemination of antibiotic resistance genes within-individual. We provide experimental evidence for how admixture can modulate virulence and metabolism. Leveraging data on the contextual microbiome, we assess how interspecies interactions can shape genetic diversity and mobile gene elements. Our study provides insights into how within-individual evolution of human skin microbes shapes their functional diversification.
Keywords: Staphylococcus epidermidis; genomic variation; metagenomics; microevolution; skin microbiome; strain diversity.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests The authors declare no competing interests.
Figures
Similar articles
-
Comprehensive genomic landscape of antibiotic resistance in Staphylococcus epidermidis.mSystems. 2024 Jun 18;9(6):e0022624. doi: 10.1128/msystems.00226-24. Epub 2024 May 10. mSystems. 2024. PMID: 38727238 Free PMC article.
-
Staphylococcus epidermidis pan-genome sequence analysis reveals diversity of skin commensal and hospital infection-associated isolates.Genome Biol. 2012 Jul 25;13(7):R64. doi: 10.1186/gb-2012-13-7-r64. Genome Biol. 2012. PMID: 22830599 Free PMC article.
-
Antagonism between Staphylococcus epidermidis and Propionibacterium acnes and its genomic basis.BMC Genomics. 2016 Feb 29;17:152. doi: 10.1186/s12864-016-2489-5. BMC Genomics. 2016. PMID: 26924200 Free PMC article.
-
Staphylococcus epidermidis and its dual lifestyle in skin health and infection.Nat Rev Microbiol. 2023 Feb;21(2):97-111. doi: 10.1038/s41579-022-00780-3. Epub 2022 Aug 30. Nat Rev Microbiol. 2023. PMID: 36042296 Free PMC article. Review.
-
Success through diversity - how Staphylococcus epidermidis establishes as a nosocomial pathogen.Int J Med Microbiol. 2010 Aug;300(6):380-6. doi: 10.1016/j.ijmm.2010.04.011. Epub 2010 May 6. Int J Med Microbiol. 2010. PMID: 20451447 Review.
Cited by
-
Skin Microbiome Variation with Cancer Progression in Human Cutaneous Squamous Cell Carcinoma.J Invest Dermatol. 2022 Oct;142(10):2773-2782.e16. doi: 10.1016/j.jid.2022.03.017. Epub 2022 Apr 4. J Invest Dermatol. 2022. PMID: 35390349 Free PMC article.
-
Comparative transcriptomic analysis of Staphylococcus epidermidis associated with periprosthetic joint infection under in vivo and in vitro conditions.Int J Med Microbiol. 2024 Jun;315:151620. doi: 10.1016/j.ijmm.2024.151620. Epub 2024 Mar 30. Int J Med Microbiol. 2024. PMID: 38579524 Free PMC article.
-
Staphylococcus epidermidis isolates from atopic or healthy skin have opposite effect on skin cells: potential implication of the AHR pathway modulation.Front Immunol. 2023 May 26;14:1098160. doi: 10.3389/fimmu.2023.1098160. eCollection 2023. Front Immunol. 2023. PMID: 37304256 Free PMC article.
-
Surface Proteins of Staphylococcus epidermidis.Front Microbiol. 2020 Jul 29;11:1829. doi: 10.3389/fmicb.2020.01829. eCollection 2020. Front Microbiol. 2020. PMID: 32849430 Free PMC article. Review.
-
Low Concentration of the Neutrophil Proteases Cathepsin G, Cathepsin B, Proteinase-3 and Metalloproteinase-9 Induce Biofilm Formation in Non-Biofilm-Forming Staphylococcus epidermidis Isolates.Int J Mol Sci. 2022 Apr 30;23(9):4992. doi: 10.3390/ijms23094992. Int J Mol Sci. 2022. PMID: 35563384 Free PMC article.
References
-
- Altschul SF, Gish W, Miller W, Myers EW, and Lipman DJ (1990). Basic local alignment search tool. J. Mol. Biol 215, 403–410. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
