

Transcriptome analyses of liver in newly-hatched chicks during the metabolic perturbation of fasting and re-feeding reveals THRSPA as the key lipogenic transcription factor
- PMID: 32005146
- PMCID: PMC6995218
- DOI: 10.1186/s12864-020-6525-0
Transcriptome analyses of liver in newly-hatched chicks during the metabolic perturbation of fasting and re-feeding reveals THRSPA as the key lipogenic transcription factor
Abstract
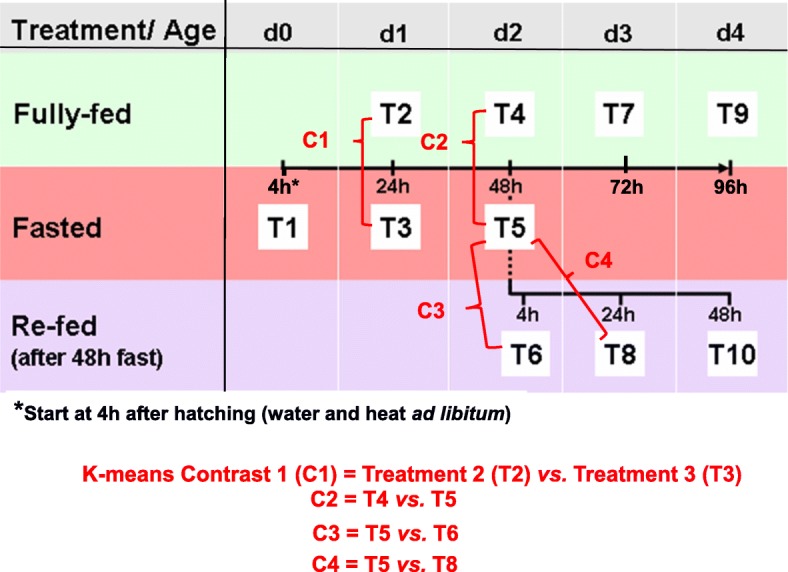
Background: The fasting-refeeding perturbation has been used extensively to reveal specific genes and metabolic pathways that control energy metabolism in the chicken. Most global transcriptional scans of the fasting-refeeding response in liver have focused on juvenile chickens that were 1, 2 or 4 weeks old. The present study was aimed at the immediate post-hatch period, in which newly-hatched chicks were subjected to fasting for 4, 24 or 48 h, then refed for 4, 24 or 48 h, and compared with a fully-fed control group at each age (D1-D4).
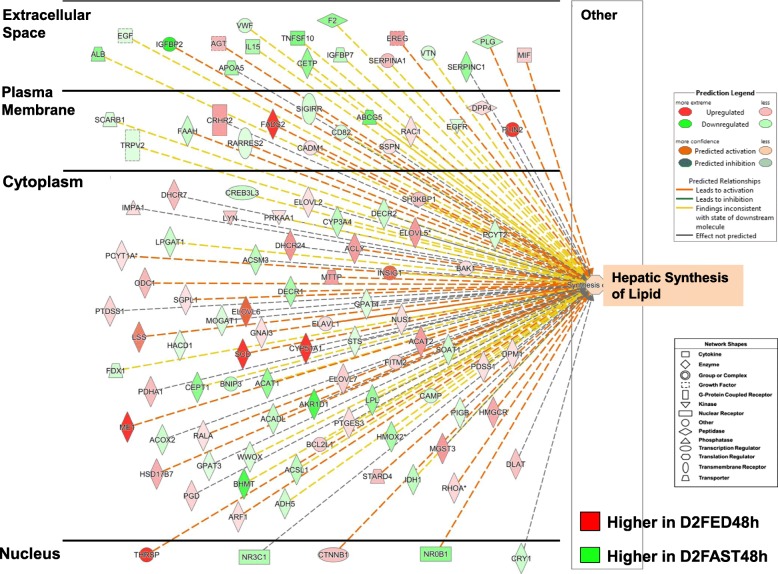
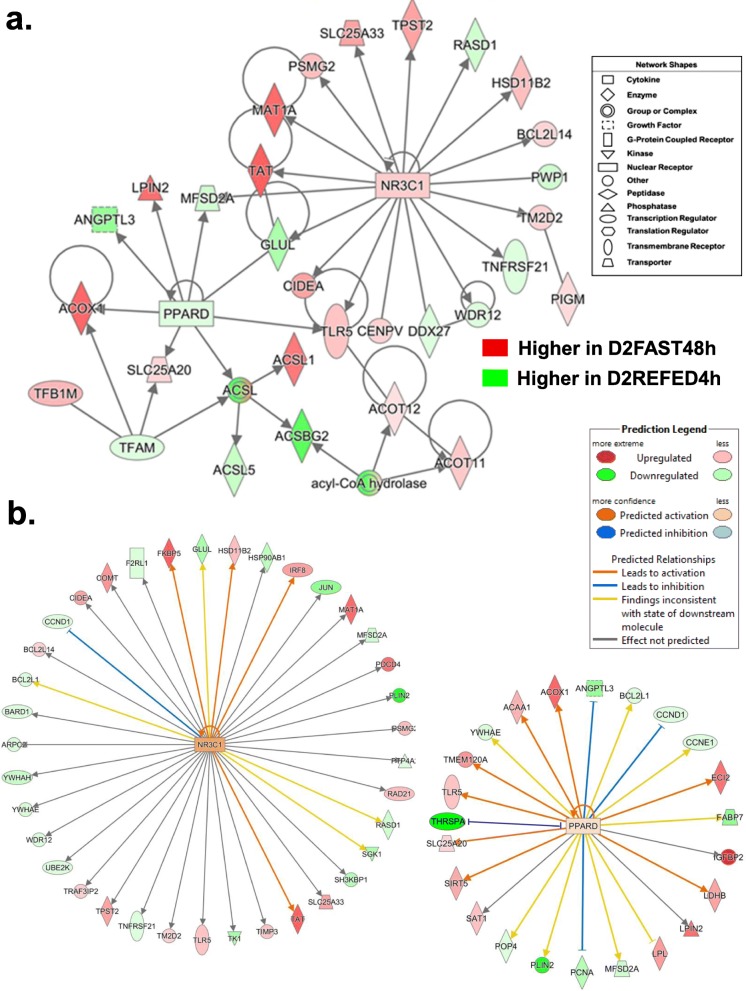
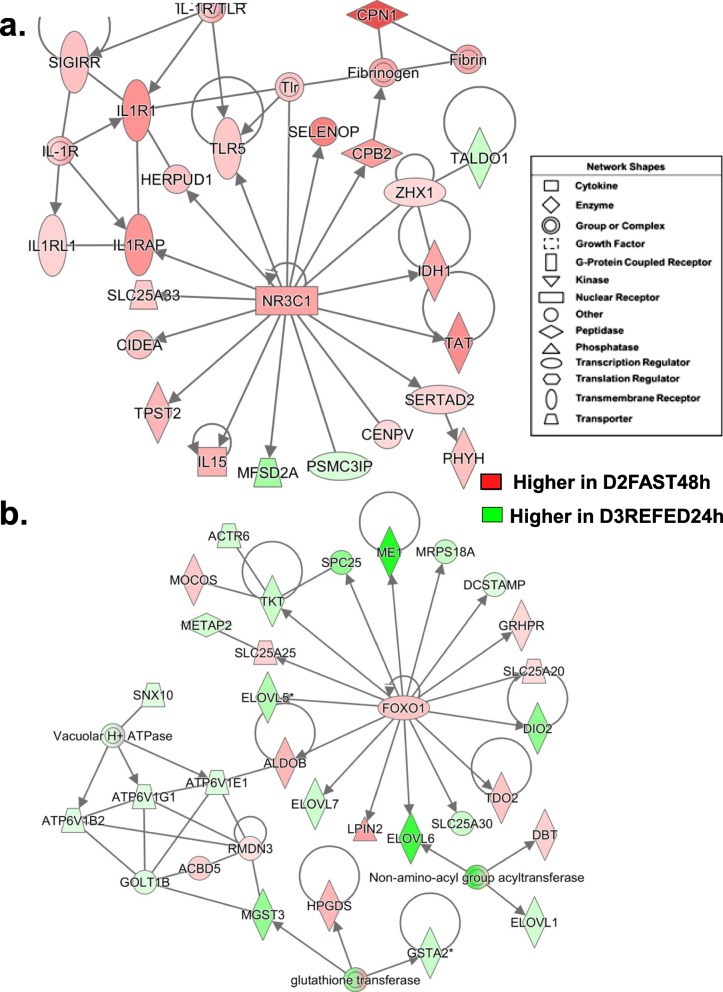
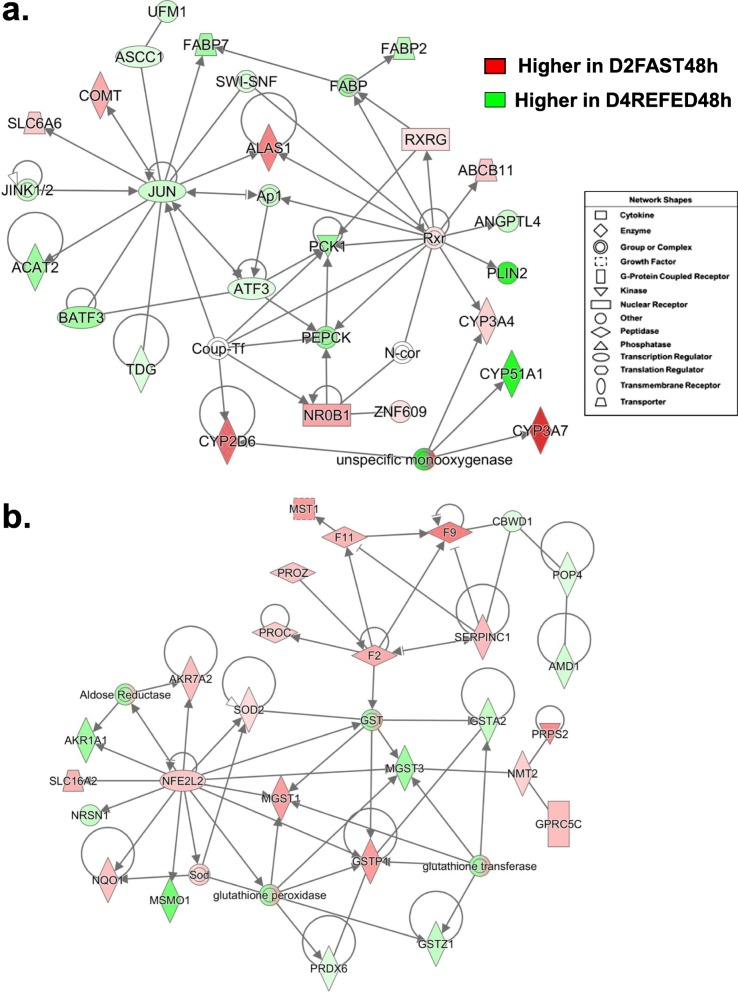
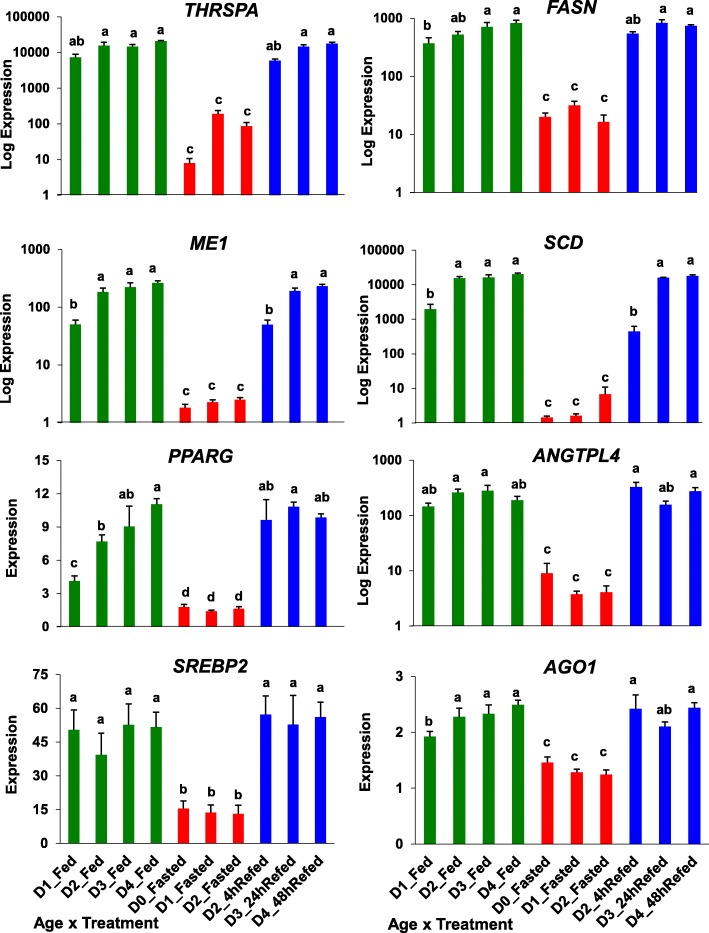
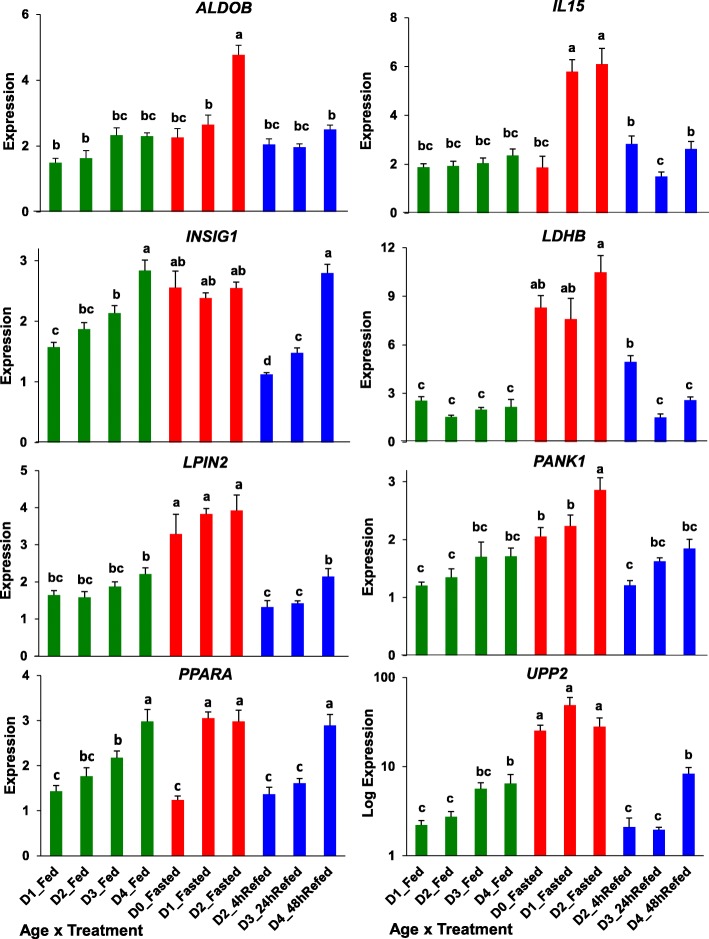
Results: Visual analysis of hepatic gene expression profiles using hierarchical and K-means clustering showed two distinct patterns, genes with higher expression during fasting and depressed expression upon refeeding and those with an opposing pattern of expression, which exhibit very low expression during fasting and more abundant expression with refeeding. Differentially-expressed genes (DEGs), identified from five prominent pair-wise contrasts of fed, fasted and refed conditions, were subjected to Ingenuity Pathway Analysis. This enabled mapping of analysis-ready (AR)-DEGs to canonical and metabolic pathways controlled by distinct gene interaction networks. The largest number of hepatic DEGs was identified by two contrasts: D2FED48h/D2FAST48h (968 genes) and D2FAST48h/D3REFED24h (1198 genes). The major genes acutely depressed by fasting and elevated upon refeeding included ANGTPL, ATPCL, DIO2, FASN, ME1, SCD, PPARG, SREBP2 and THRSPA-a primary lipogenic transcription factor. In contrast, major lipolytic genes were up-regulated by fasting or down-regulated after refeeding, including ALDOB, IL-15, LDHB, LPIN2, NFE2L2, NR3C1, NR0B1, PANK1, PPARA, SERTAD2 and UPP2.
Conclusions: Transcriptional profiling of liver during fasting/re-feeding of newly-hatched chicks revealed several highly-expressed upstream regulators, which enable the metabolic switch from fasted (lipolytic/gluconeogenic) to fed or refed (lipogenic/thermogenic) states. This rapid homeorhetic shift of whole-body metabolism from a catabolic-fasting state to an anabolic-fed state appears precisely orchestrated by a small number of ligand-activated transcription factors that provide either a fasting-lipolytic state (PPARA, NR3C1, NFE2L2, SERTAD2, FOX01, NR0B1, RXR) or a fully-fed and refed lipogenic/thermogenic state (THRSPA, SREBF2, PPARG, PPARD, JUN, ATF3, CTNNB1). THRSPA has emerged as the key transcriptional regulator that drives lipogenesis and thermogenesis in hatchling chicks, as shown here in fed and re-fed states.
Keywords: Gene interaction networks; Homeorhesis; Lipid metabolism; Lipogenesis; Lipolysis; Metabolic switch; Reciprocal inhibition/activation; Spot 14 (THRSPA); THRSP paralogs; Target genes; Thermogenesis; Up-stream regulators; ying-yang metabolic regulation.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures






References
-
- Cogburn LA, Morgan R, Burnside J. Expressed sequence tags, DNA chip technology and gene expression profiling. In: Muir WM, Aggrey SE, editors. Poultry Genetics, Breeding and Biotechnology. Wallingford: CABI Publishing; 2003. pp. 629–646.
-
- Cogburn LA, Wang X, Carré W, Rejto L, Aggrey SE, Duclos MJ, Simon J, Porter TE. Functional genomics in chickens: development of integrated-systems microarrays for transcriptional profiling and discovery of regulatory pathways. Comp Funct Genom. 2004;5:253–261. doi: 10.1002/cfg.402. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
