

DSP p.(Thr2104Glnfs*12) variant presents variably with early onset severe arrhythmias and left ventricular cardiomyopathy
- PMID: 32005173
- PMCID: PMC6995042
- DOI: 10.1186/s12881-020-0955-z
DSP p.(Thr2104Glnfs*12) variant presents variably with early onset severe arrhythmias and left ventricular cardiomyopathy
Abstract
Background: Dilated cardiomyopathy (DCM) is a condition characterized by dilatation and systolic dysfunction of the left ventricle in the absence of severe coronary artery disease or abnormal loading conditions. Mutations in the titin (TTN) and lamin A/C (LMNA) genes are the two most significant contributors in familial DCM. Previously mutations in the desmoplakin (DSP) gene have been associated with arrhythmogenic right ventricular cardiomyopathy (ARVC) and more recently with DCM.
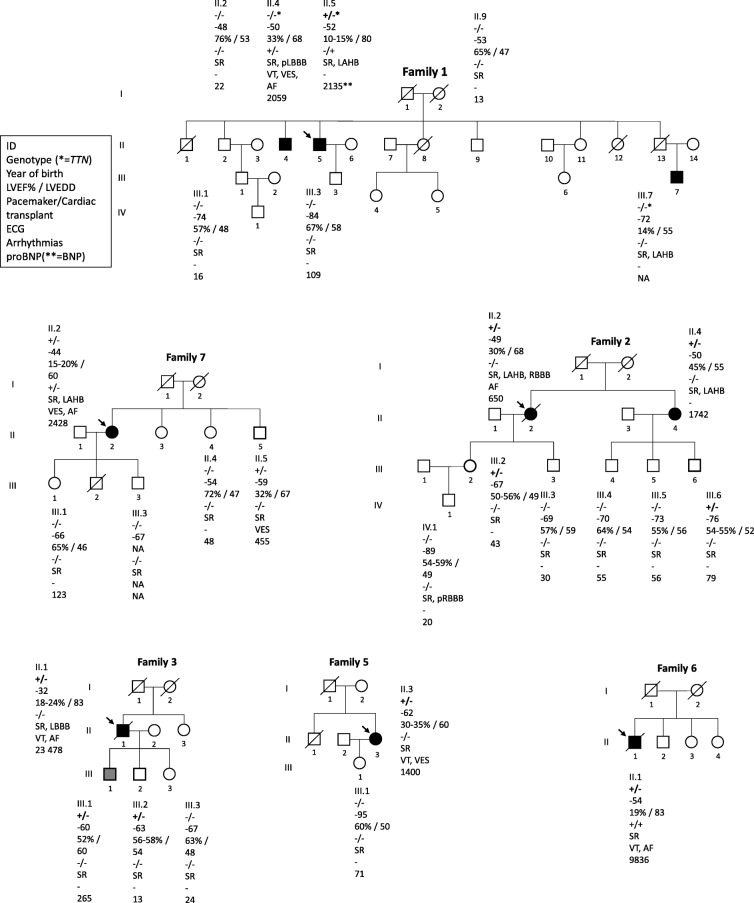
Methods: We describe the cardiac phenotype related to a DSP mutation which was identified in ten unrelated Finnish index patients using next-generation sequencing. Sanger sequencing was used to verify the presence of this DSP variant in the probands' relatives. Medical records were obtained, and clinical evaluation was performed.
Results: We identified DSP c.6310delA, p.(Thr2104Glnfs*12) variant in 17 individuals of which 11 (65%) fulfilled the DCM diagnostic criteria. This pathogenic variant presented with left ventricular dilatation, dysfunction and major ventricular arrhythmias. Two patients showed late gadolinium enhancement (LGE) and myocardial edema on cardiac magnetic resonance imaging (MRI) that may suggest inflammatory process at myocardium.
Conclusions: The patients diagnosed with DCM showed an arrhythmogenic phenotype as well as SCD at young age supporting the recently proposed concept of arrhythmogenic cardiomyopathy. This study also demonstrates relatively low penetrance of truncating DSP variant in the probands' family members by the age of 40. Further studies are needed to elucidate the possible relations between myocardial inflammation and pathogenic DSP variants.
Keywords: Arrhythmogenic cardiomyopathy; Cardiomyopathies; DSP; Dilated cardiomyopathy; Mutation; desmoplakin.
Conflict of interest statement
Minor conflict of interest: TPA, SM, JK are co-founders and TKK, TPA, SM, JK are full-time employees of Blueprint Genetics, which offers genetic diagnostic services.
Figures
References
-
- Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, et al. Classification of the cardiomyopathies: A position statement from the european society of cardiology working group on myocardial and pericardial diseases. Eur Heart J. 2008;29(2):270–276. doi: 10.1093/eurheartj/ehm342. - DOI - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Miscellaneous
