

ATR-16 syndrome: mechanisms linking monosomy to phenotype
- PMID: 32005695
- PMCID: PMC7279195
- DOI: 10.1136/jmedgenet-2019-106528
ATR-16 syndrome: mechanisms linking monosomy to phenotype
Abstract
Background: Deletions removing 100s-1000s kb of DNA, and variable numbers of poorly characterised genes, are often found in patients with a wide range of developmental abnormalities. In such cases, understanding the contribution of the deletion to an individual's clinical phenotype is challenging.
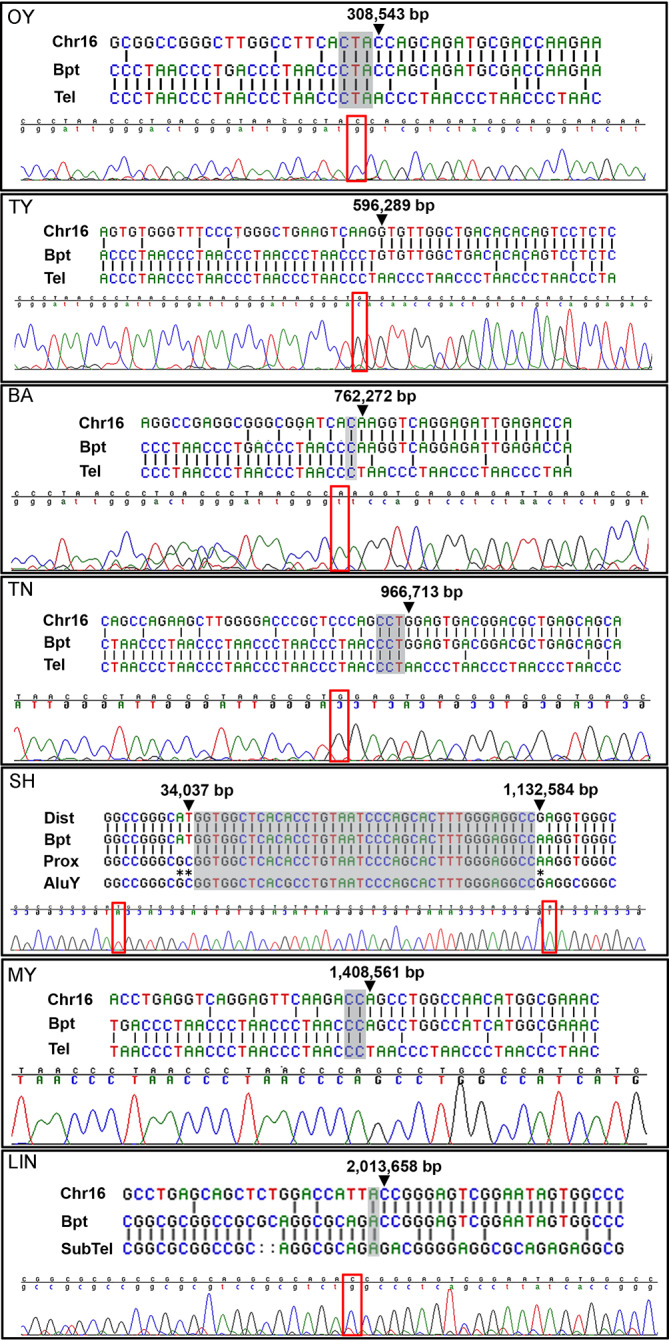
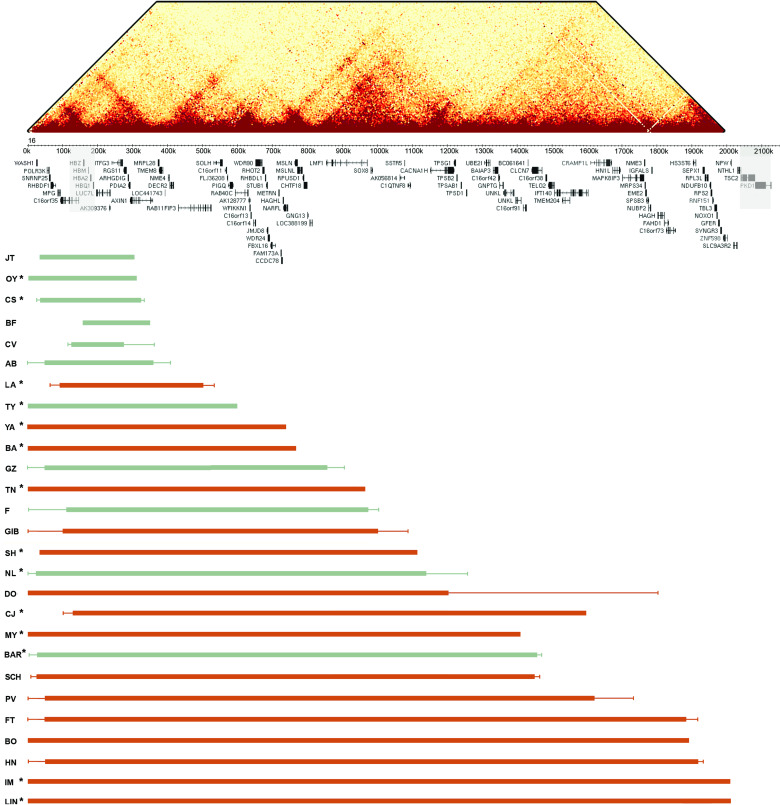
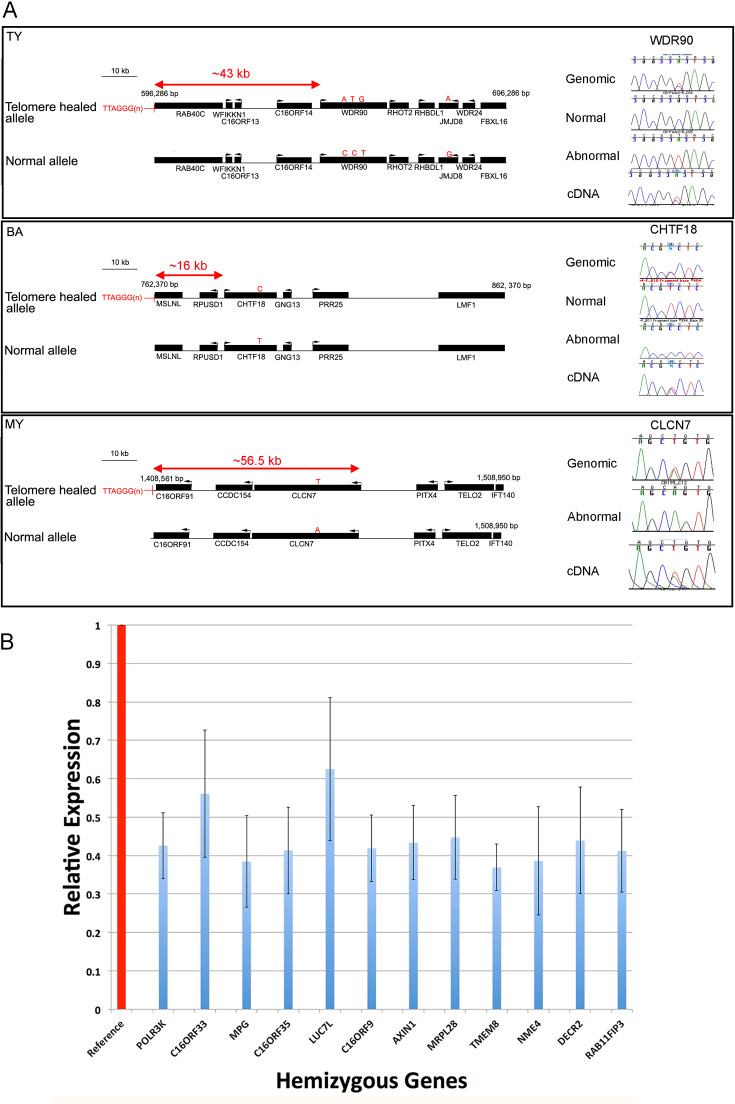
Methods: Here, as an example of this common phenomenon, we analysed 41 patients with simple deletions of ~177 to ~2000 kb affecting one allele of the well-characterised, gene dense, distal region of chromosome 16 (16p13.3), referred to as ATR-16 syndrome. We characterised deletion extents and screened for genetic background effects, telomere position effect and compensatory upregulation of hemizygous genes.
Results: We find the risk of developmental and neurological abnormalities arises from much smaller distal chromosome 16 deletions (~400 kb) than previously reported. Beyond this, the severity of ATR-16 syndrome increases with deletion size, but there is no evidence that critical regions determine the developmental abnormalities associated with this disorder. Surprisingly, we find no evidence of telomere position effect or compensatory upregulation of hemizygous genes; however, genetic background effects substantially modify phenotypic abnormalities.
Conclusions: Using ATR-16 as a general model of disorders caused by CNVs, we show the degree to which individuals with contiguous gene syndromes are affected is not simply related to the number of genes deleted but depends on their genetic background. We also show there is no critical region defining the degree of phenotypic abnormalities in ATR-16 syndrome and this has important implications for genetic counselling.
Keywords: ATR16; CNV; developmental delay; thalassemia.
© Author(s) (or their employer(s)) 2020. Re-use permitted under CC BY. Published by BMJ.
Conflict of interest statement
Competing interests: None declared.
Figures
References
-
- Kaminsky EB, Kaul V, Paschall J, Church DM, Bunke B, Kunig D, Moreno-De-Luca D, Moreno-De-Luca A, Mulle JG, Warren ST, Richard G, Compton JG, Fuller AE, Gliem TJ, Huang S, Collinson MN, Beal SJ, Ackley T, Pickering DL, Golden DM, Aston E, Whitby H, Shetty S, Rossi MR, Rudd MK, South ST, Brothman AR, Sanger WG, Iyer RK, Crolla JA, Thorland EC, Aradhya S, Ledbetter DH, Martin CL. An evidence-based approach to establish the functional and clinical significance of copy number variants in intellectual and developmental disabilities. Genet Med 2011;13:777–84. 10.1097/GIM.0b013e31822c79f9 - DOI - PMC - PubMed
-
- Wilkie AO, Buckle VJ, Harris PC, Lamb J, Barton NJ, Reeders ST, Lindenbaum RH, Nicholls RD, Barrow M, Bethlenfalvay NC, Hutz MH, Tolmie JL, Weatherall DJ, Higgs DR. Clinical features and molecular analysis of the alpha thalassemia/mental retardation syndromes. I. cases due to deletions involving chromosome band 16p13.3. Am J Hum Genet 1990;46:1112–26. - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous