

Human and mouse essentiality screens as a resource for disease gene discovery
- PMID: 32005800
- PMCID: PMC6994715
- DOI: 10.1038/s41467-020-14284-2
Human and mouse essentiality screens as a resource for disease gene discovery
Abstract
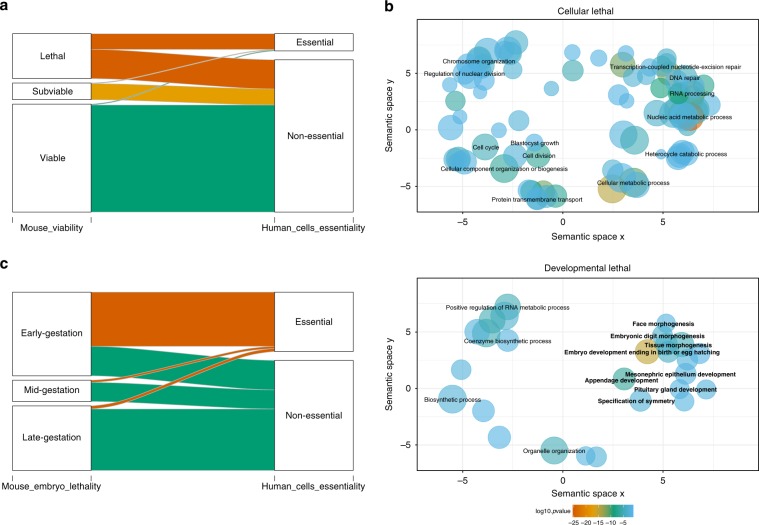
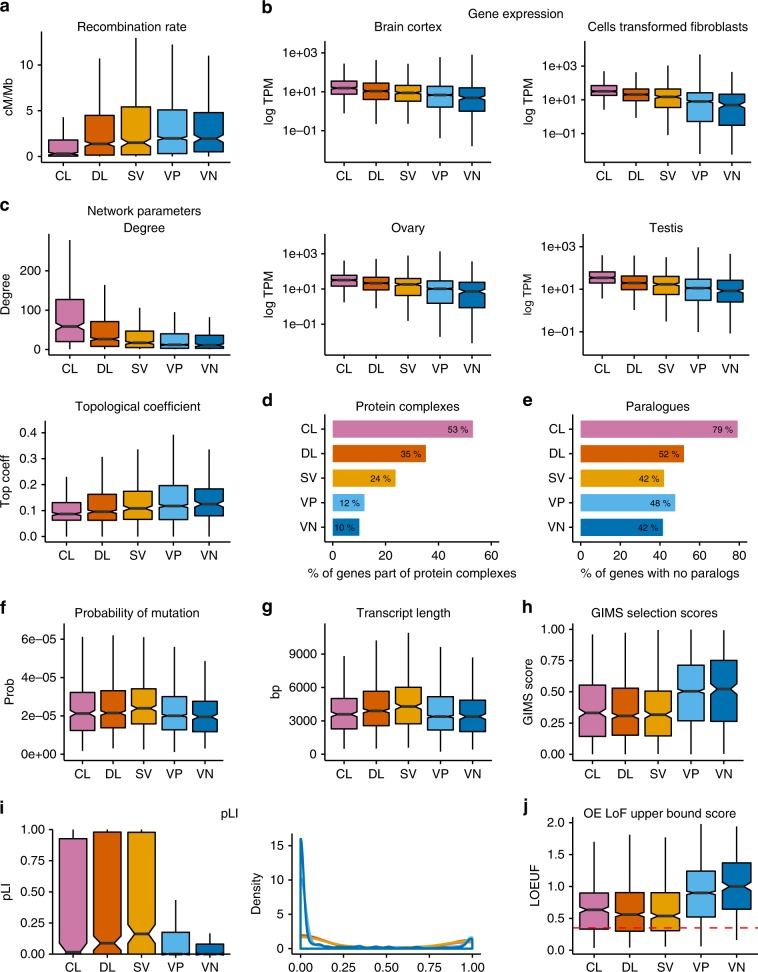
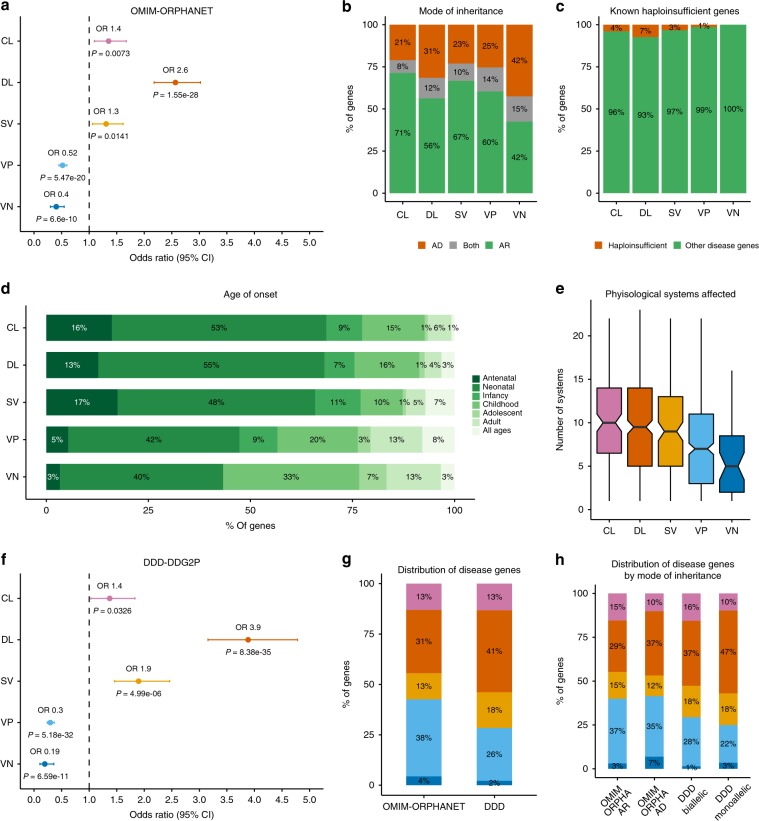
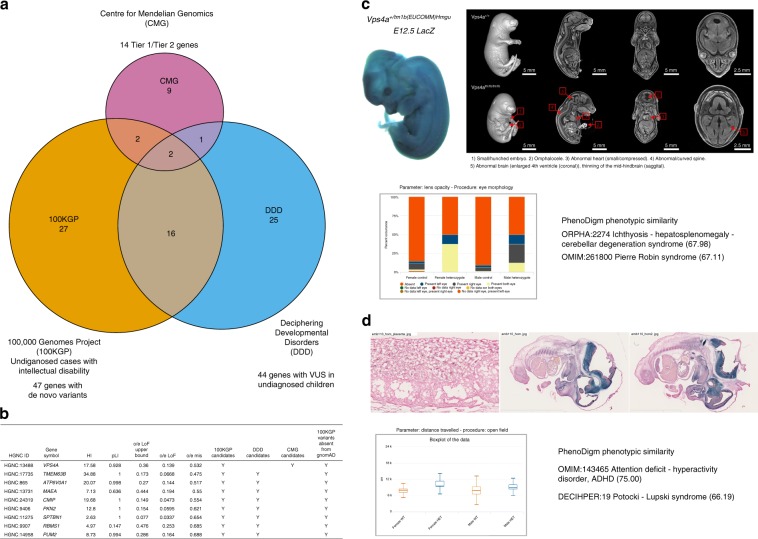
The identification of causal variants in sequencing studies remains a considerable challenge that can be partially addressed by new gene-specific knowledge. Here, we integrate measures of how essential a gene is to supporting life, as inferred from viability and phenotyping screens performed on knockout mice by the International Mouse Phenotyping Consortium and essentiality screens carried out on human cell lines. We propose a cross-species gene classification across the Full Spectrum of Intolerance to Loss-of-function (FUSIL) and demonstrate that genes in five mutually exclusive FUSIL categories have differing biological properties. Most notably, Mendelian disease genes, particularly those associated with developmental disorders, are highly overrepresented among genes non-essential for cell survival but required for organism development. After screening developmental disorder cases from three independent disease sequencing consortia, we identify potentially pathogenic variants in genes not previously associated with rare diseases. We therefore propose FUSIL as an efficient approach for disease gene discovery.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
- UM1 HG008900/HG/NHGRI NIH HHS/United States
- UM1 HG006504/HG/NHGRI NIH HHS/United States
- MC_UP_1502/1/MRC_/Medical Research Council/United Kingdom
- UM1 HG006542/HG/NHGRI NIH HHS/United States
- UM1 OD023221/OD/NIH HHS/United States
- MC_U142684172/MRC_/Medical Research Council/United Kingdom
- UM1 HG006370/HG/NHGRI NIH HHS/United States
- UM1 HG006493/HG/NHGRI NIH HHS/United States
- U54 HG006370/HG/NHGRI NIH HHS/United States
- MC_U142684171/MRC_/Medical Research Council/United Kingdom
- U54 HG006364/HG/NHGRI NIH HHS/United States
- UM1 HG006348/HG/NHGRI NIH HHS/United States
- U42 OD011174/OD/NIH HHS/United States
- U42 OD011175/OD/NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
- MR/S006753/1/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
