

Tracking hematopoietic stem cells and their progeny using whole-genome sequencing
- PMID: 32007478
- PMCID: PMC7118367
- DOI: 10.1016/j.exphem.2020.01.004
Tracking hematopoietic stem cells and their progeny using whole-genome sequencing
Abstract
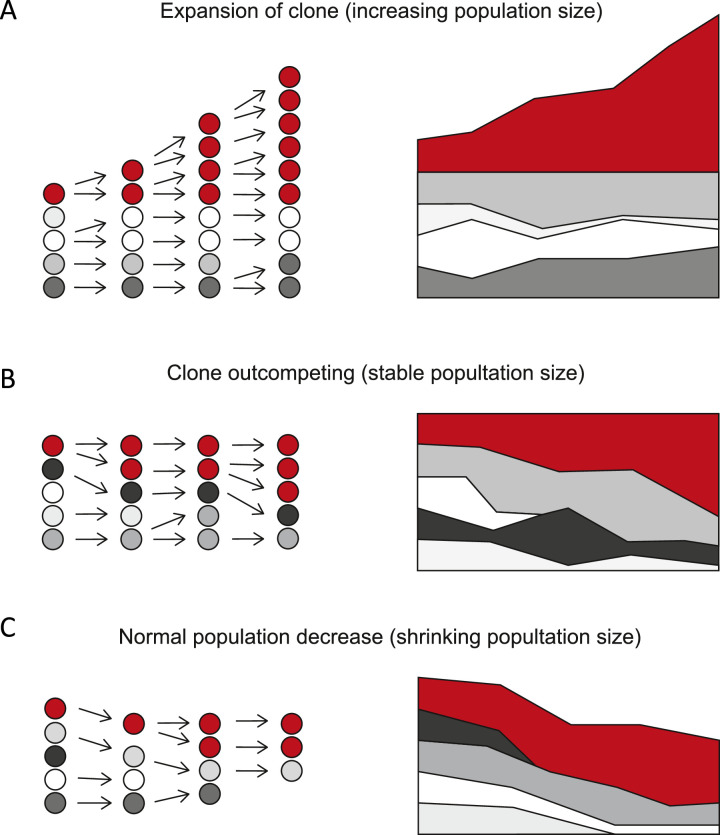
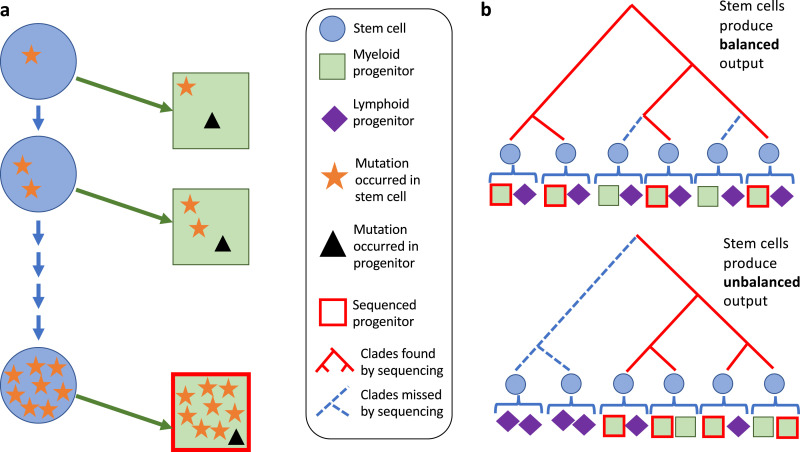
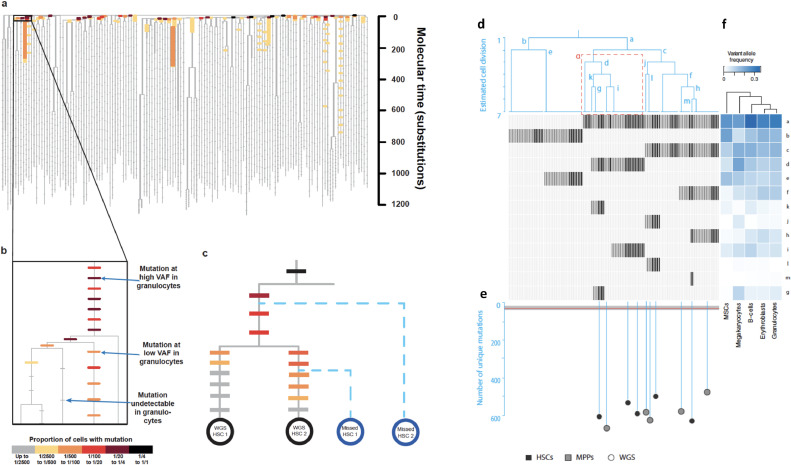
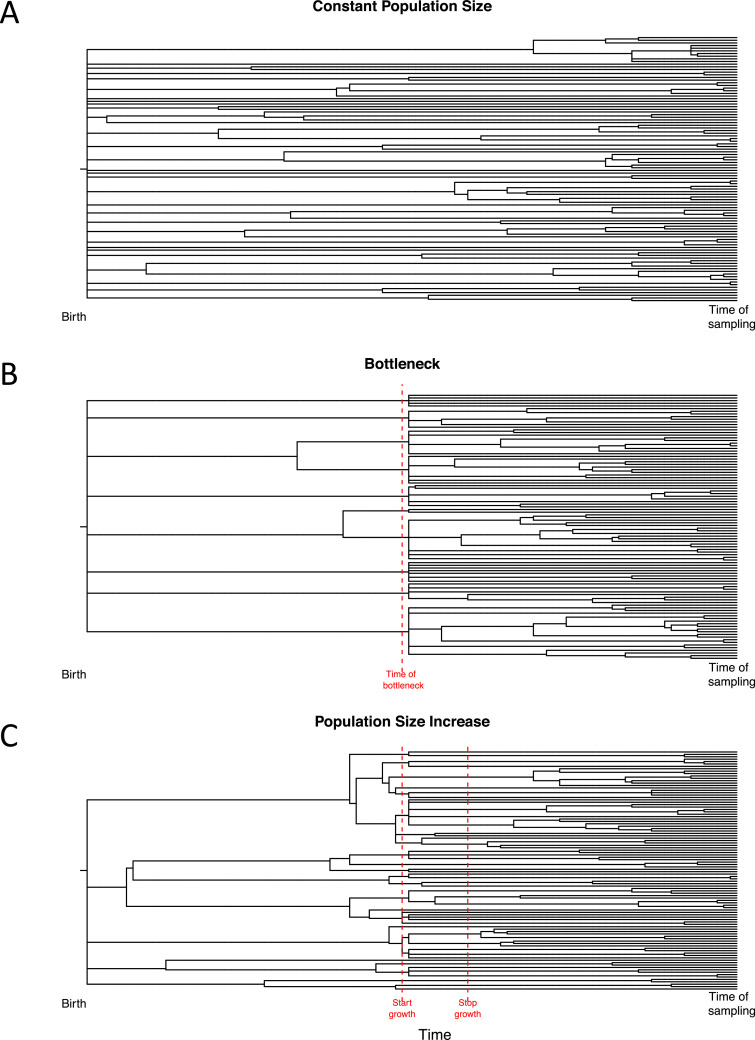
Despite decades of progress in our understanding of hematopoiesis through the study of animal models and transplantation in humans, investigating physiological human hematopoiesis directly has remained challenging. Questions on the clonal structure of the human hematopoietic stem cell (HSC) pool, such as "how many HSCs are there?" and "do all HSC clones actively produce all blood cell types in equal proportions?" remain open. These questions have inherent value for understanding normal human physiology, but also directly inform our comprehension of the process by which the system is subverted to drive diseases of the blood, in particular blood cancers and bone marrow failure syndromes. The critical link between normal and abnormal hematopoiesis is perhaps best illustrated by the recent discovery of clonal hematopoiesis in healthy people with no abnormal blood parameters. In such individuals, large clones derived from single cells are present and are dominant relative to their normal counterparts, but their presence does not necessitate abnormal blood cell production. Intriguingly, however, these individuals are also at a significantly greater risk of developing leukemias and of cardiovascular events, underscoring the importance of understanding how blood stem cell clones compete against each other.
Copyright © 2020 ISEH -- Society for Hematology and Stem Cells. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Ayachi S, Buscarlet M, Busque L. 60 years of clonal hematopoiesis research: From X-chromosome inactivation studies to the identification of driver mutations. Exp Hematol. 2020;83:2–11. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
