

A Novel Homozygous Variant in the Fork-Head-Associated Domain of Polynucleotide Kinase Phosphatase in a Patient Affected by Late-Onset Ataxia With Oculomotor Apraxia Type 4
- PMID: 32010037
- PMCID: PMC6974581
- DOI: 10.3389/fneur.2019.01331
A Novel Homozygous Variant in the Fork-Head-Associated Domain of Polynucleotide Kinase Phosphatase in a Patient Affected by Late-Onset Ataxia With Oculomotor Apraxia Type 4
Abstract
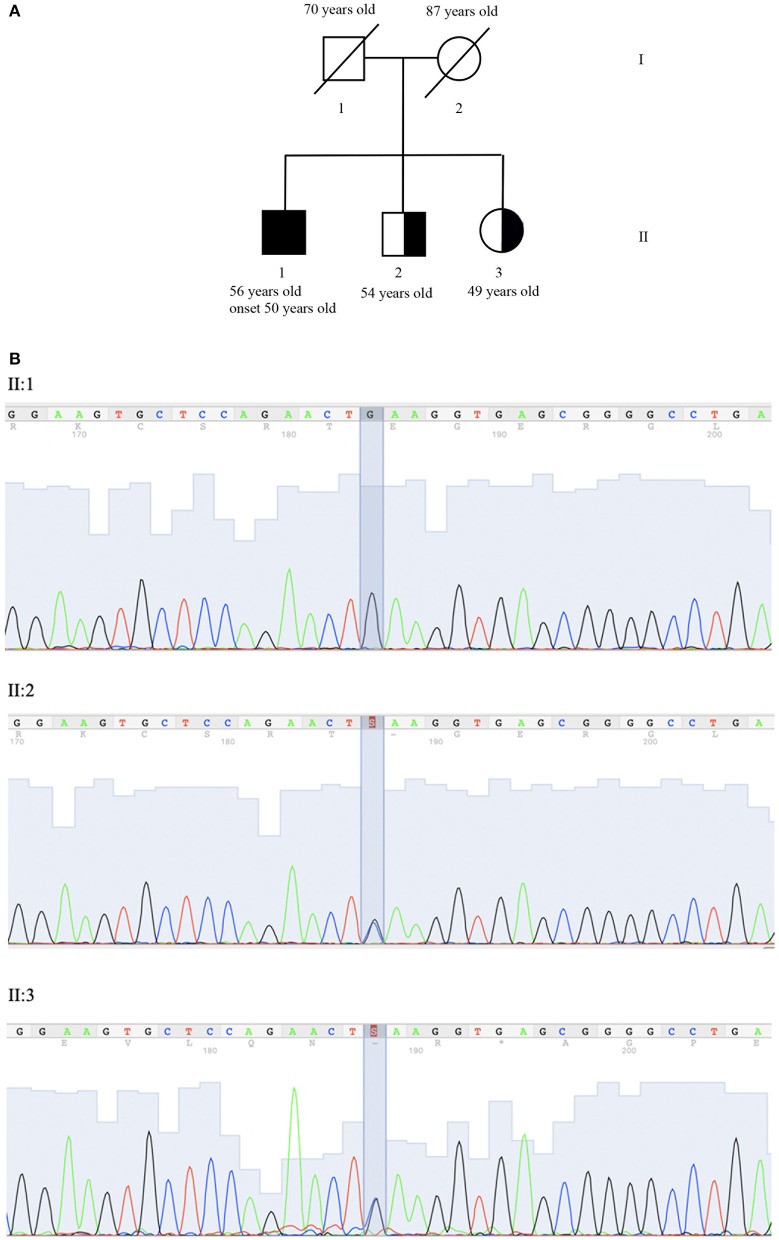
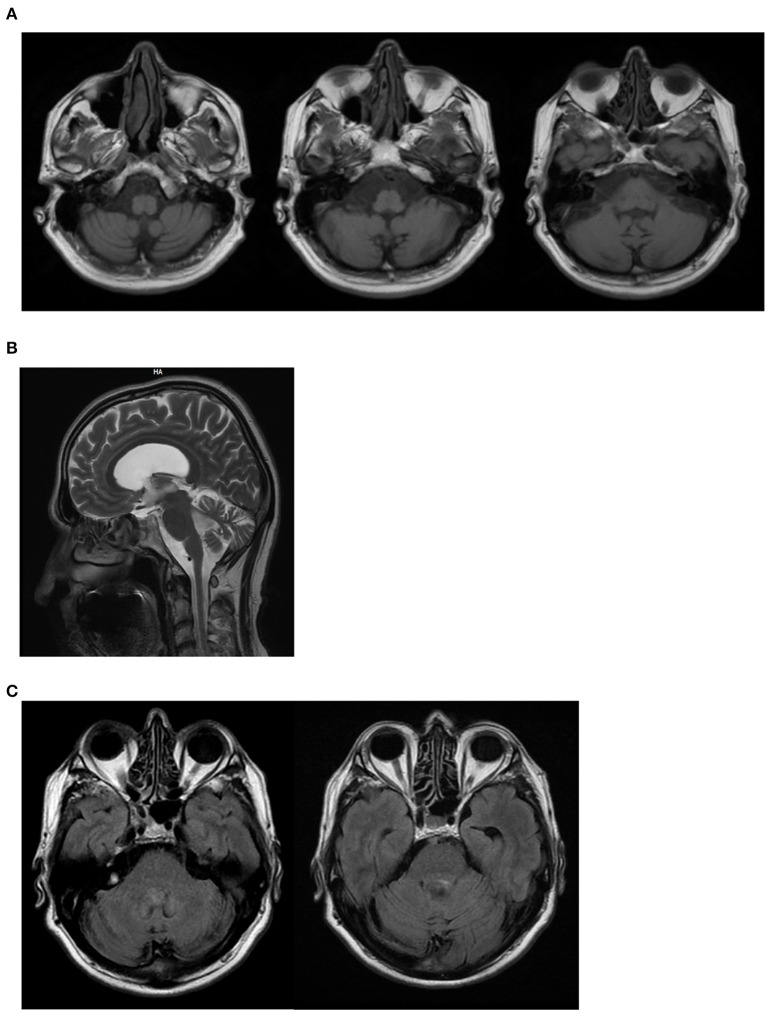
Ataxia with oculomotor apraxia (AOA) is a clinical syndrome featuring a group of genetic diseases including at least four separate autosomal-recessive cerebellar ataxias. All these disorders are due to altered genes involved in DNA repair. AOA type 4 (AOA4) is caused by mutations in DNA repair factor polynucleotide kinase phosphatase (PNKP), which encodes for a DNA processing enzyme also involved in other syndromes featured by microcephaly or neurodegeneration. To date, only a few AOA4 patients have been reported worldwide. All these patients are homozygous or compound heterozygous carriers for mutations in the kinase domain of PNKP. In this report, we describe a 56 years old patient affected by AOA4 characterized by ataxia, polyneuropathy, oculomotor apraxia, and cognitive impairment with the absence of dystonia. The disease is characterized by a very late onset (50 years) when compared with other AOA4 patients described so far (median age of onset at 4 years). In this proband, Clinical Exome Analysis through Next Generation Sequencing (NGS) consisting of 4,800 genes, identified the PNKP homozygous mutation p.Gln50Glu. This variant, classified as a likely pathogenic variant according to American College of Medical Genetics (ACMG) guidelines, does not involve the kinase domain but falls in the fork-head-associated (FHA) domain. So far, mutations in such a domain were reported to associate only with a pure seizure syndrome without the classic AOA4 features. Therefore, this is the first report of patients carrying a mutation of the FHA domain within the PNKP gene which expresses the clinical phenotype known as the AOA4 syndrome and the lack of any seizure activity. Further studies are required to investigate specifically the significance of various mutations within the FHA domain, and it would be worth to correlate these variants with the age of onset of the AOA4 syndrome.
Keywords: PNKP gene; ataxia with oculomotor apraxia; clinical exome; late onset; neurogenetics.
Copyright © 2020 Campopiano, Ferese, Buttari, Femiano, Centonze, Fornai, Biagioni, Chiaravalloti, Magnani, Giardina, Ruzzo and Gambardella.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources