

DeepWAS: Multivariate genotype-phenotype associations by directly integrating regulatory information using deep learning
- PMID: 32012148
- PMCID: PMC7043350
- DOI: 10.1371/journal.pcbi.1007616
DeepWAS: Multivariate genotype-phenotype associations by directly integrating regulatory information using deep learning
Abstract
Genome-wide association studies (GWAS) identify genetic variants associated with traits or diseases. GWAS never directly link variants to regulatory mechanisms. Instead, the functional annotation of variants is typically inferred by post hoc analyses. A specific class of deep learning-based methods allows for the prediction of regulatory effects per variant on several cell type-specific chromatin features. We here describe "DeepWAS", a new approach that integrates these regulatory effect predictions of single variants into a multivariate GWAS setting. Thereby, single variants associated with a trait or disease are directly coupled to their impact on a chromatin feature in a cell type. Up to 61 regulatory SNPs, called dSNPs, were associated with multiple sclerosis (MS, 4,888 cases and 10,395 controls), major depressive disorder (MDD, 1,475 cases and 2,144 controls), and height (5,974 individuals). These variants were mainly non-coding and reached at least nominal significance in classical GWAS. The prediction accuracy was higher for DeepWAS than for classical GWAS models for 91% of the genome-wide significant, MS-specific dSNPs. DSNPs were enriched in public or cohort-matched expression and methylation quantitative trait loci and we demonstrated the potential of DeepWAS to generate testable functional hypotheses based on genotype data alone. DeepWAS is available at https://github.com/cellmapslab/DeepWAS.
Conflict of interest statement
The authors declare that no competing interests exist.
Figures

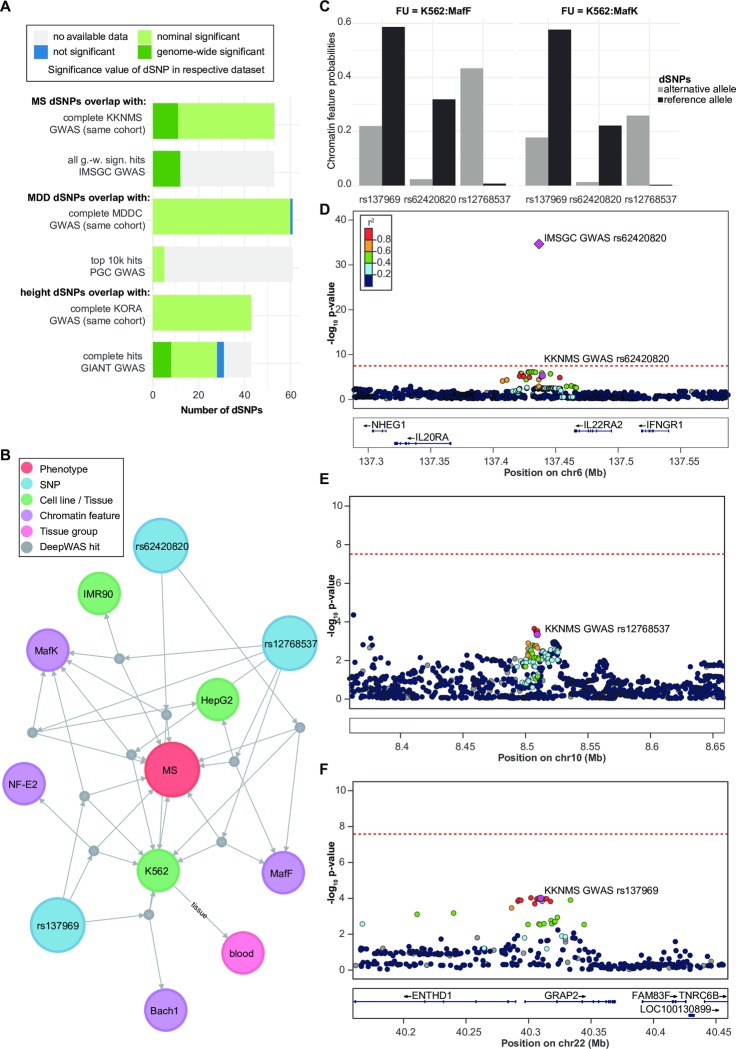
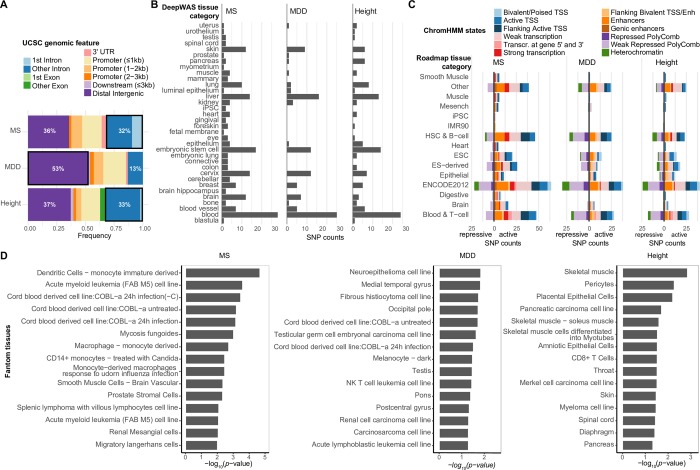

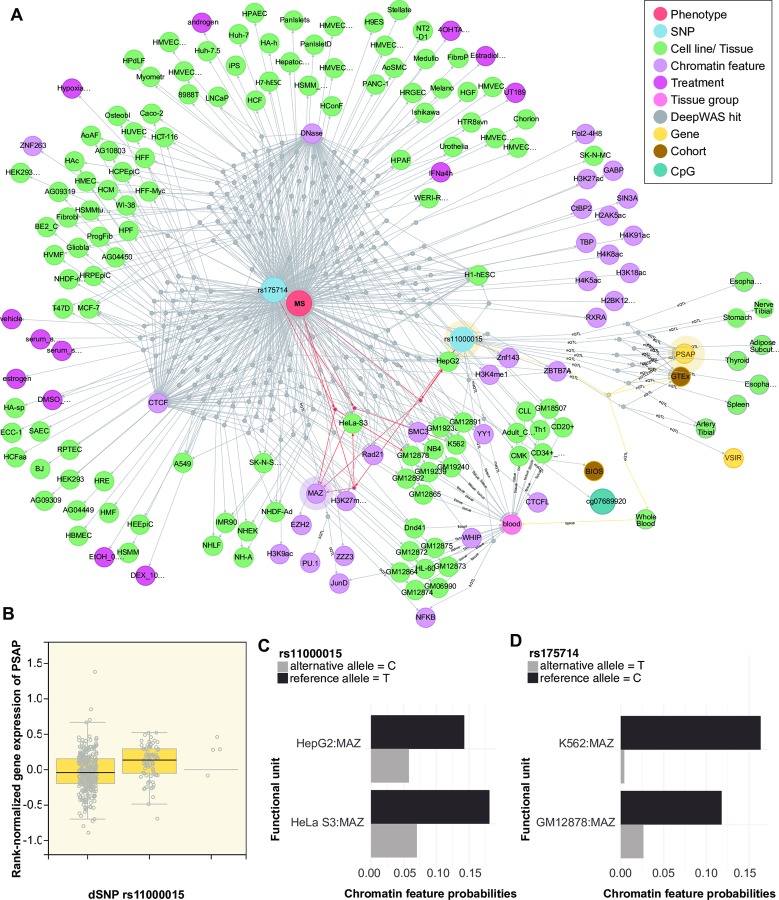
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
