

From reads to insight: a hitchhiker's guide to ATAC-seq data analysis
- PMID: 32014034
- PMCID: PMC6996192
- DOI: 10.1186/s13059-020-1929-3
From reads to insight: a hitchhiker's guide to ATAC-seq data analysis
Abstract
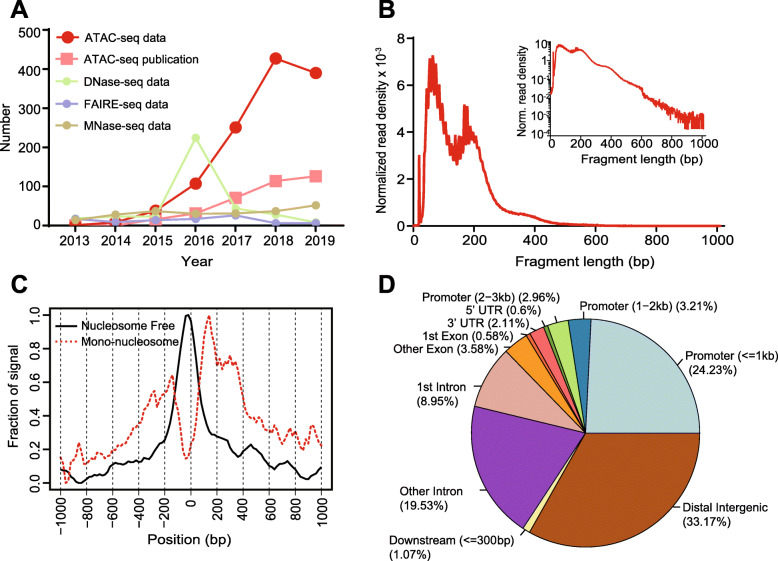
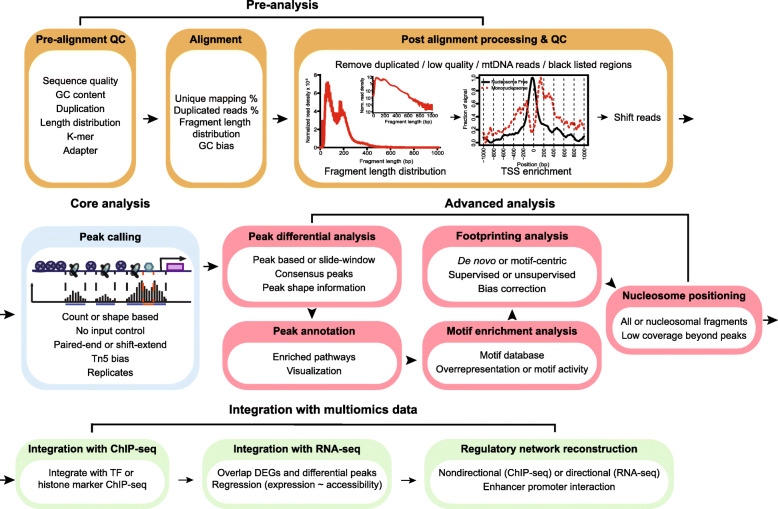
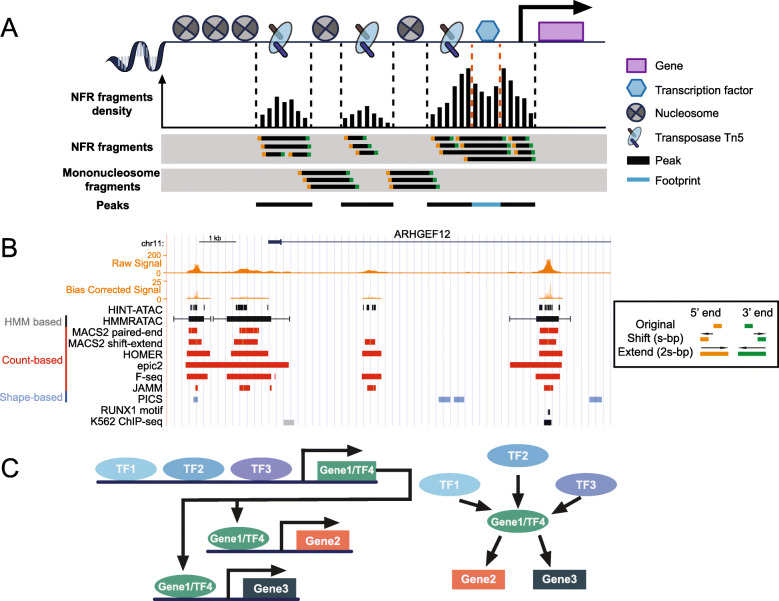
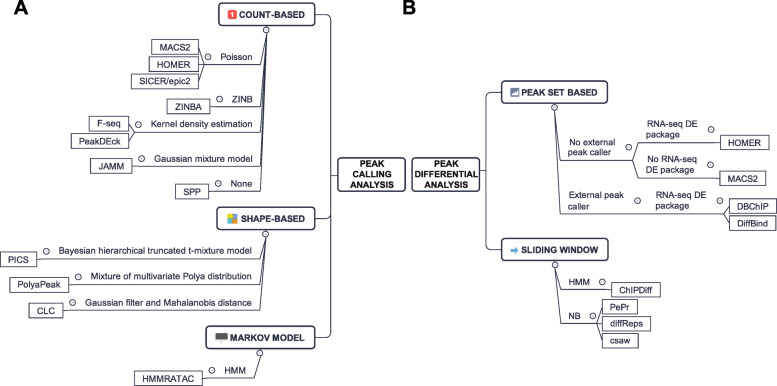
Assay of Transposase Accessible Chromatin sequencing (ATAC-seq) is widely used in studying chromatin biology, but a comprehensive review of the analysis tools has not been completed yet. Here, we discuss the major steps in ATAC-seq data analysis, including pre-analysis (quality check and alignment), core analysis (peak calling), and advanced analysis (peak differential analysis and annotation, motif enrichment, footprinting, and nucleosome position analysis). We also review the reconstruction of transcriptional regulatory networks with multiomics data and highlight the current challenges of each step. Finally, we describe the potential of single-cell ATAC-seq and highlight the necessity of developing ATAC-seq specific analysis tools to obtain biologically meaningful insights.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
