

Discovery of several thousand highly diverse circular DNA viruses
- PMID: 32014111
- PMCID: PMC7000223
- DOI: 10.7554/eLife.51971
Discovery of several thousand highly diverse circular DNA viruses
Abstract
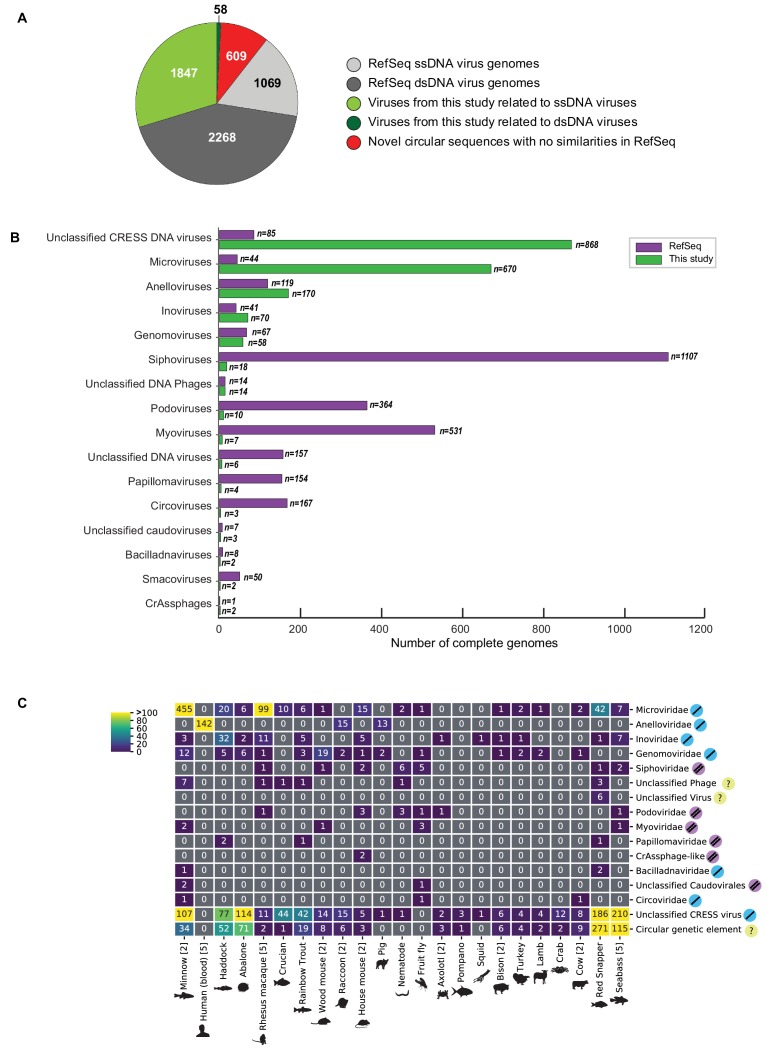
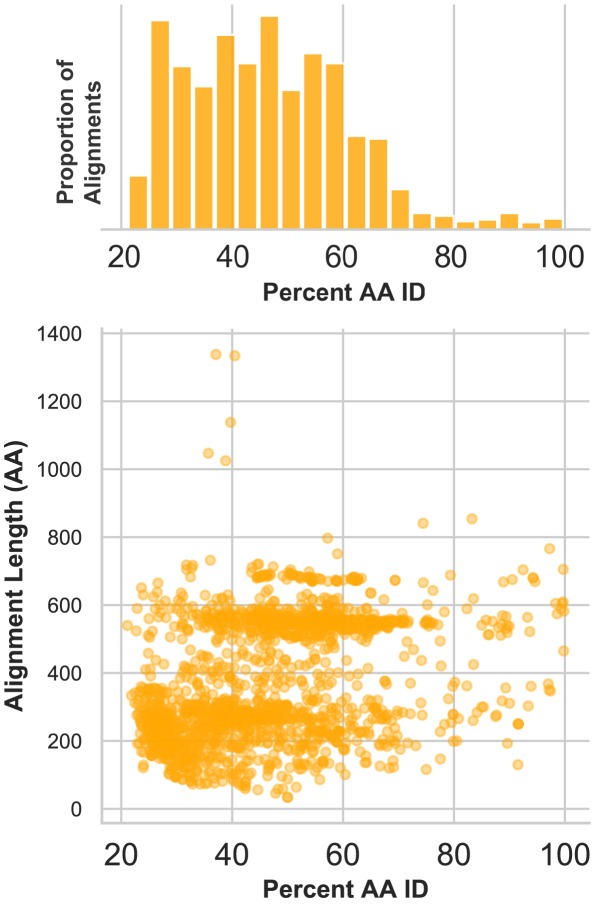
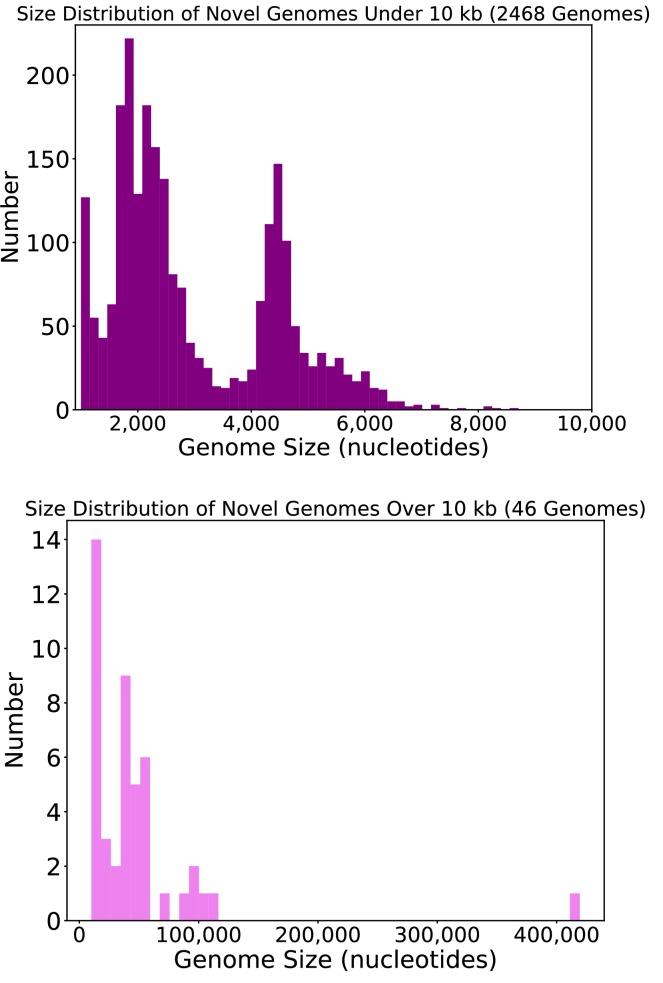
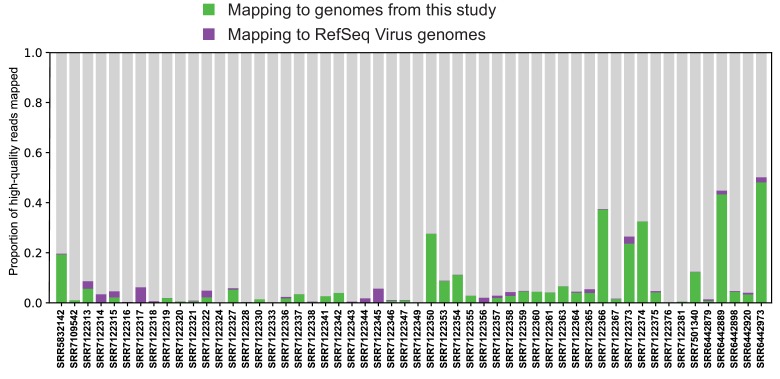
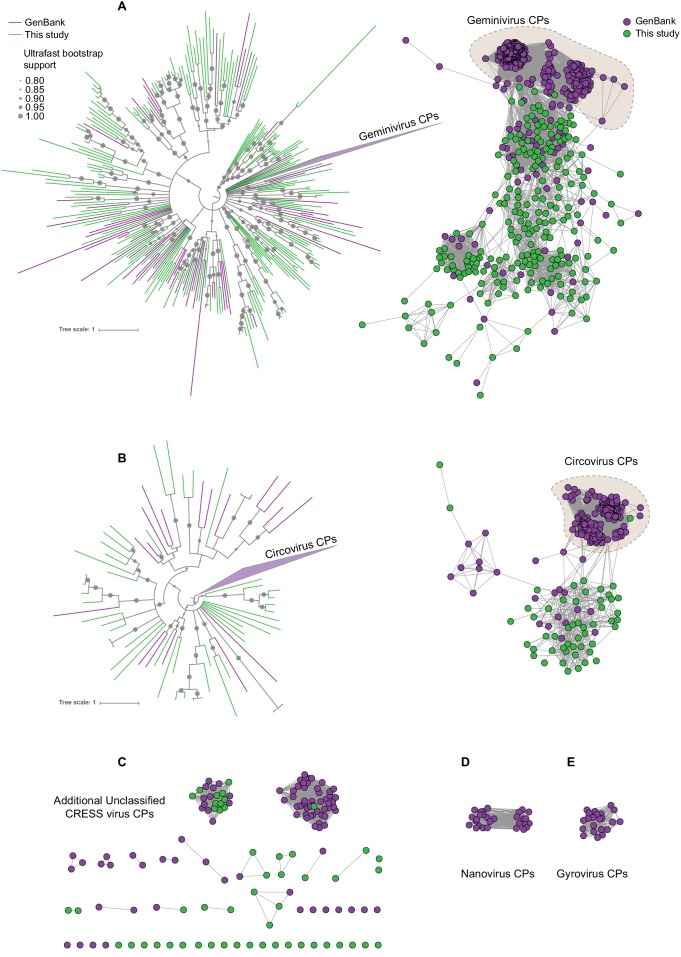
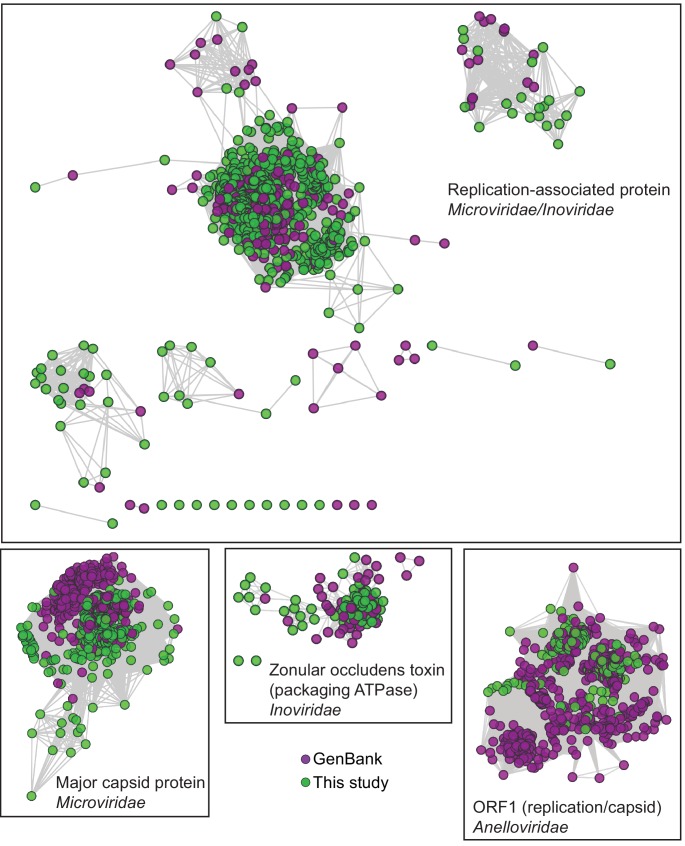
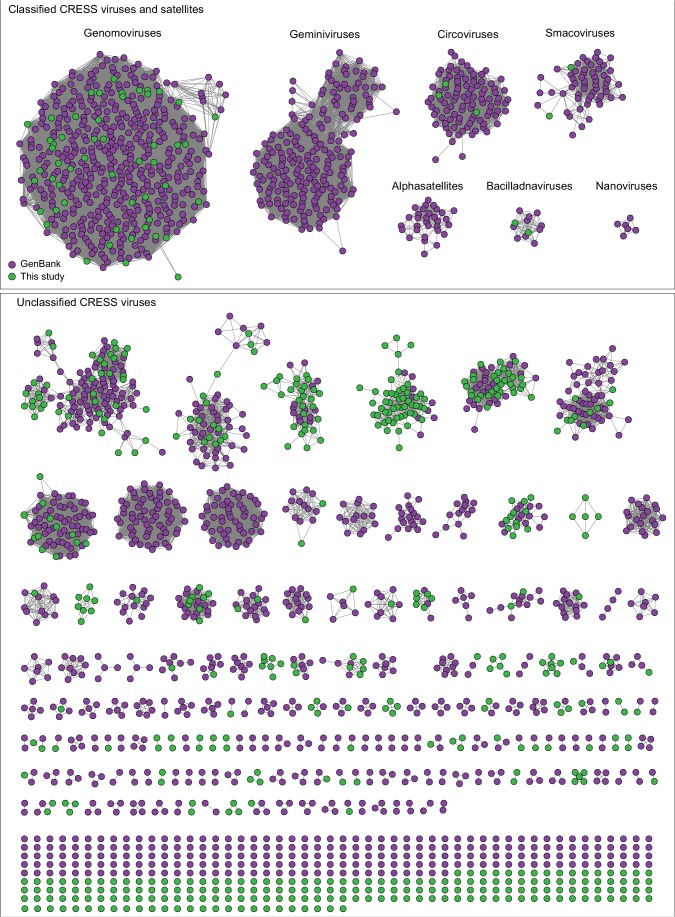
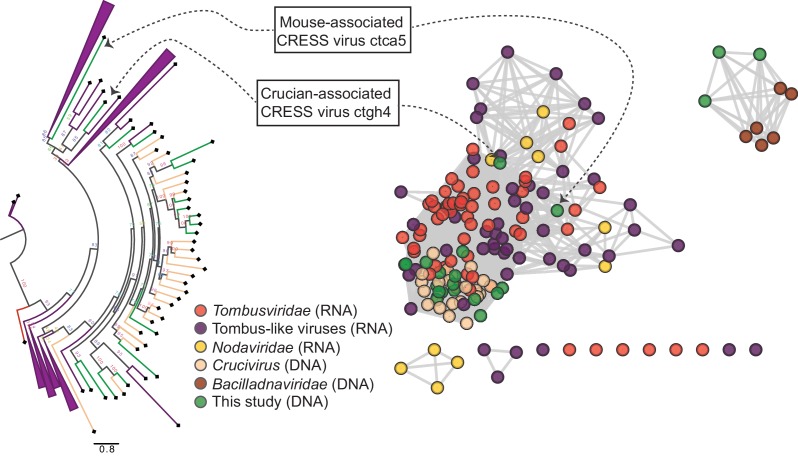
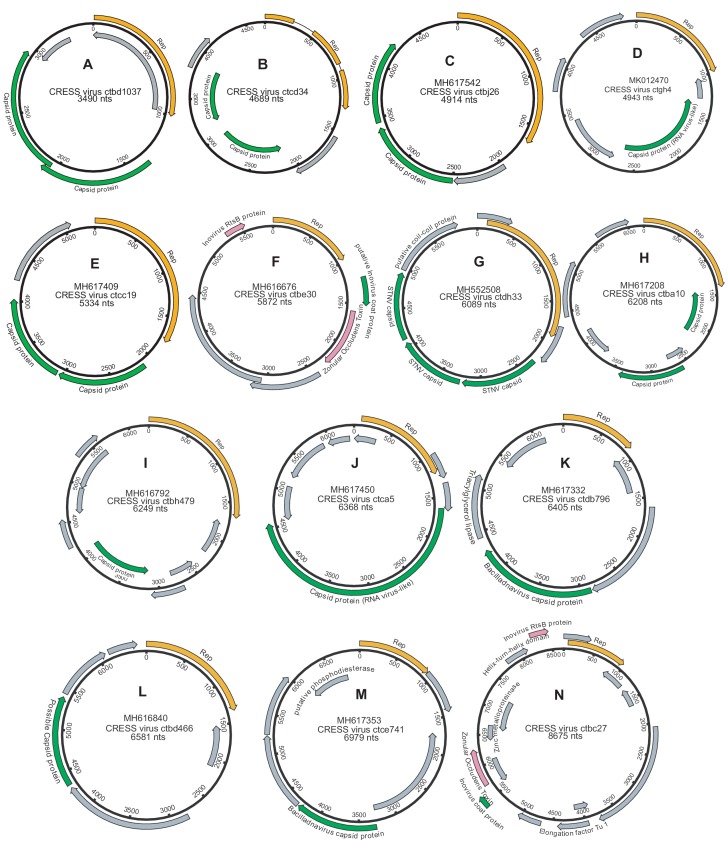
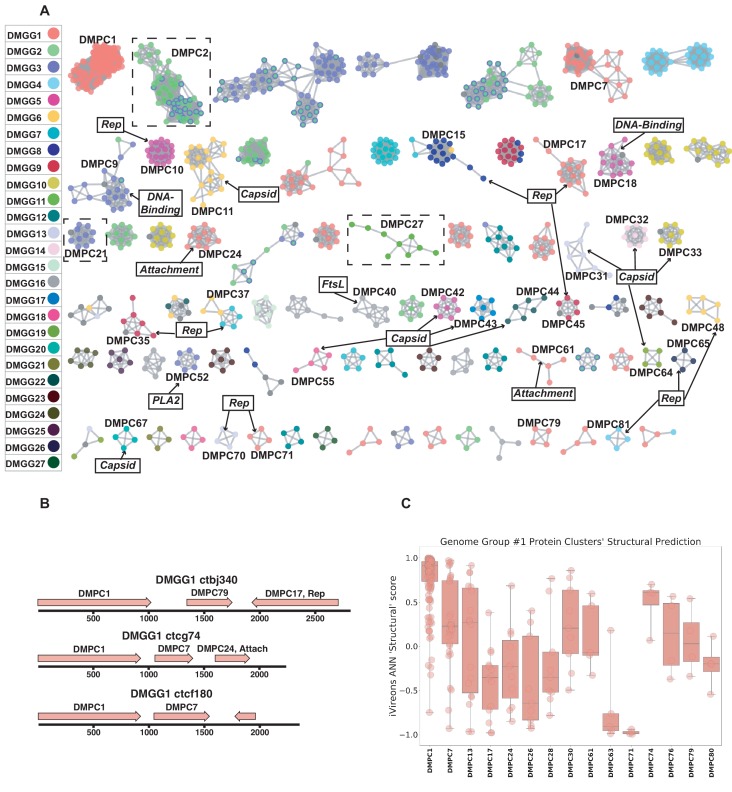
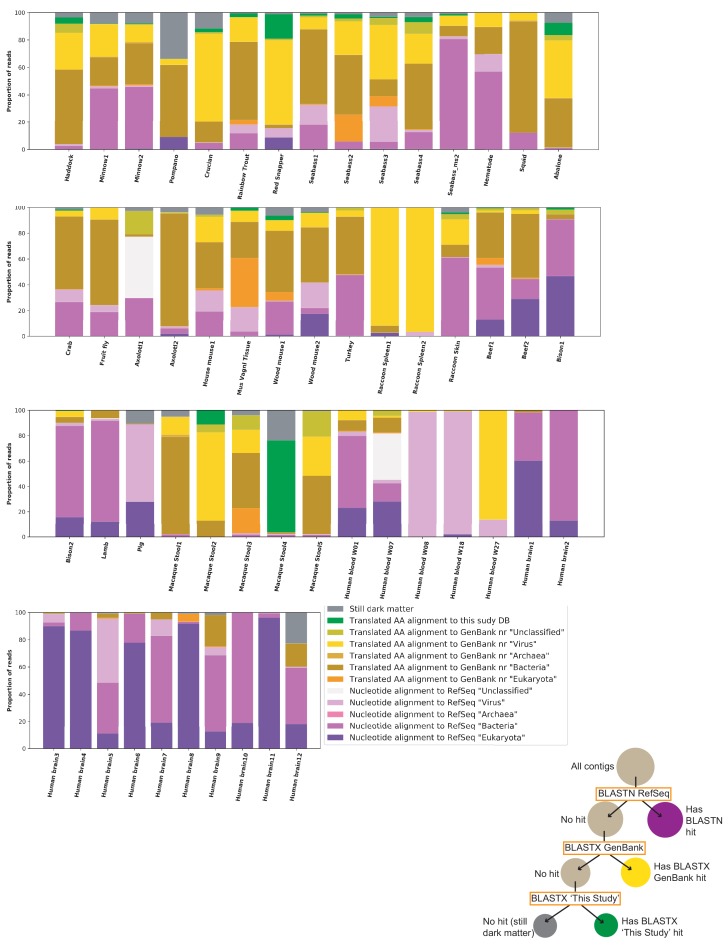
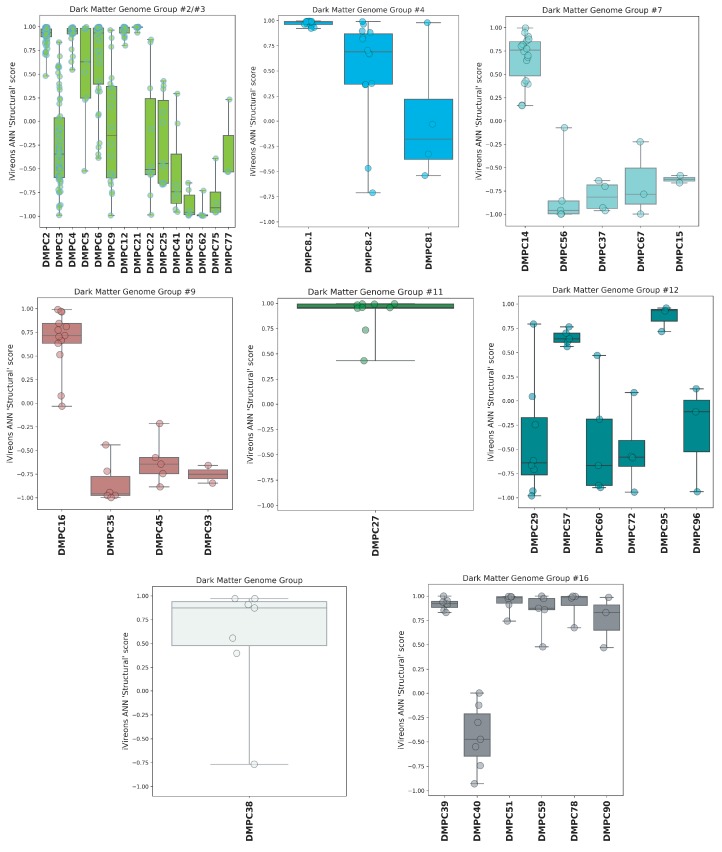
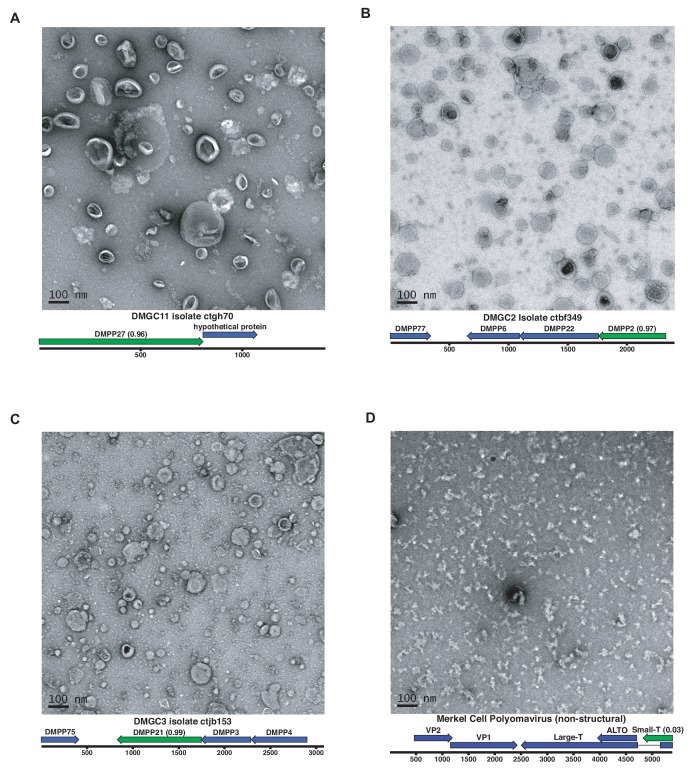
Although millions of distinct virus species likely exist, only approximately 9000 are catalogued in GenBank's RefSeq database. We selectively enriched for the genomes of circular DNA viruses in over 70 animal samples, ranging from nematodes to human tissue specimens. A bioinformatics pipeline, Cenote-Taker, was developed to automatically annotate over 2500 complete genomes in a GenBank-compliant format. The new genomes belong to dozens of established and emerging viral families. Some appear to be the result of previously undescribed recombination events between ssDNA and ssRNA viruses. In addition, hundreds of circular DNA elements that do not encode any discernable similarities to previously characterized sequences were identified. To characterize these 'dark matter' sequences, we used an artificial neural network to identify candidate viral capsid proteins, several of which formed virus-like particles when expressed in culture. These data further the understanding of viral sequence diversity and allow for high throughput documentation of the virosphere.
Keywords: evolutionary biology; infectious disease; metagenomics; microbiology; microbiome; viral evolution; virus.
Conflict of interest statement
MT, DP, NW, BS, AP, GS, YP, SK, PP, DM, PM, JW, BM, JB, SR, BR, JD, BT, OP, JT, SR, BB, MS, AV, AS, CB No competing interests declared
Figures


References
-
- Asplund M, Kjartansdóttir KR, Mollerup S, Vinner L, Fridholm H, Herrera JAR, Friis-Nielsen J, Hansen TA, Jensen RH, Nielsen IB, Richter SR, Rey-Iglesia A, Matey-Hernandez ML, Alquezar-Planas DE, Olsen PVS, Sicheritz-Pontén T, Willerslev E, Lund O, Brunak S, Mourier T, Nielsen LP, Izarzugaza JMG, Hansen AJ. Contaminating viral sequences in high-throughput sequencing viromics: a linkage study of 700 sequencing libraries. Clinical Microbiology and Infection. 2019;25:1277–1285. doi: 10.1016/j.cmi.2019.04.028. - DOI - PubMed
-
- Bin Jang H, Bolduc B, Zablocki O, Kuhn JH, Roux S, Adriaenssens EM, Brister JR, Kropinski AM, Krupovic M, Lavigne R, Turner D, Sullivan MB. Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nature Biotechnology. 2019;37:632–639. doi: 10.1038/s41587-019-0100-8. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
