

Epigenetic Mechanisms in Leukemias and Lymphomas
- PMID: 32014848
- PMCID: PMC7706582
- DOI: 10.1101/cshperspect.a034959
Epigenetic Mechanisms in Leukemias and Lymphomas
Abstract
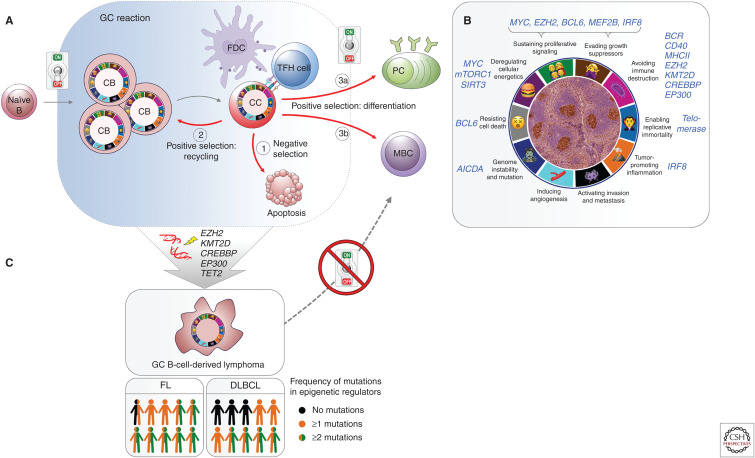
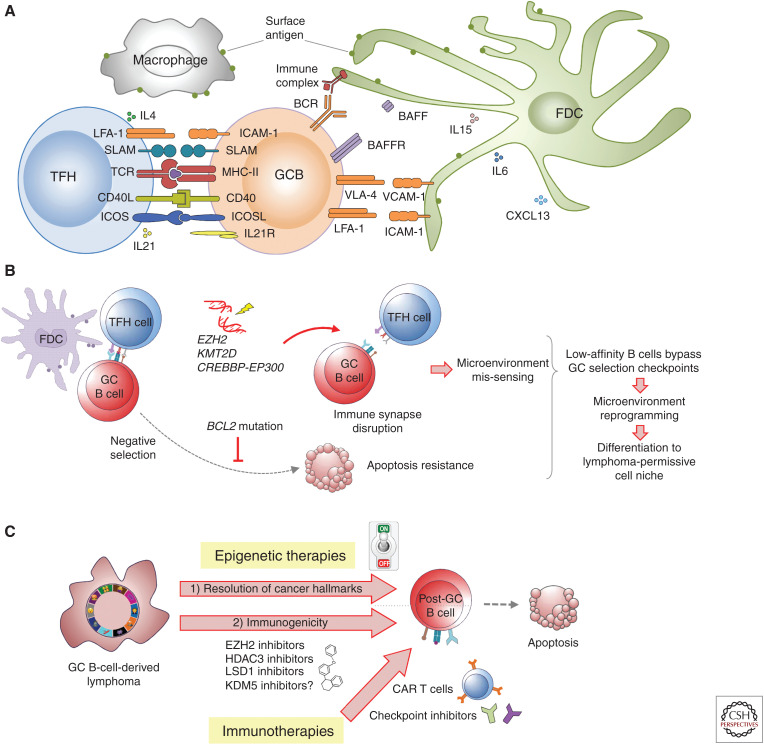
Although we are just beginning to understand the mechanisms that regulate the epigenome, aberrant epigenetic programming has already emerged as a hallmark of hematologic malignancies including acute myeloid leukemia (AML) and B-cell lymphomas. Although these diseases arise from the hematopoietic system, the epigenetic mechanisms that drive these malignancies are quite different. Yet, in all of these tumors, somatic mutations in transcription factors and epigenetic modifiers are the most commonly mutated set of genes and result in multilayered disruption of the epigenome. Myeloid and lymphoid neoplasms generally manifest epigenetic allele diversity, which contributes to tumor cell population fitness regardless of the underlying genetics. Epigenetic therapies are emerging as one of the most promising new approaches for these patients. However, effective targeting of the epigenome must consider the need to restore the various layers of epigenetic marks, appropriate biological end points, and specificity of therapeutic agents to truly realize the potential of this modality.
Copyright © 2020 Cold Spring Harbor Laboratory Press; all rights reserved.
Figures
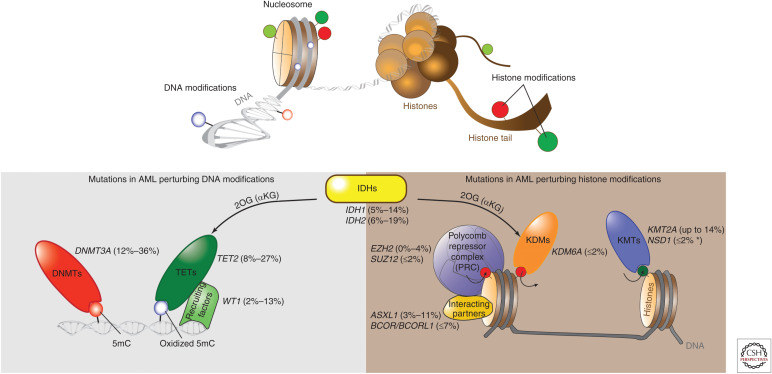
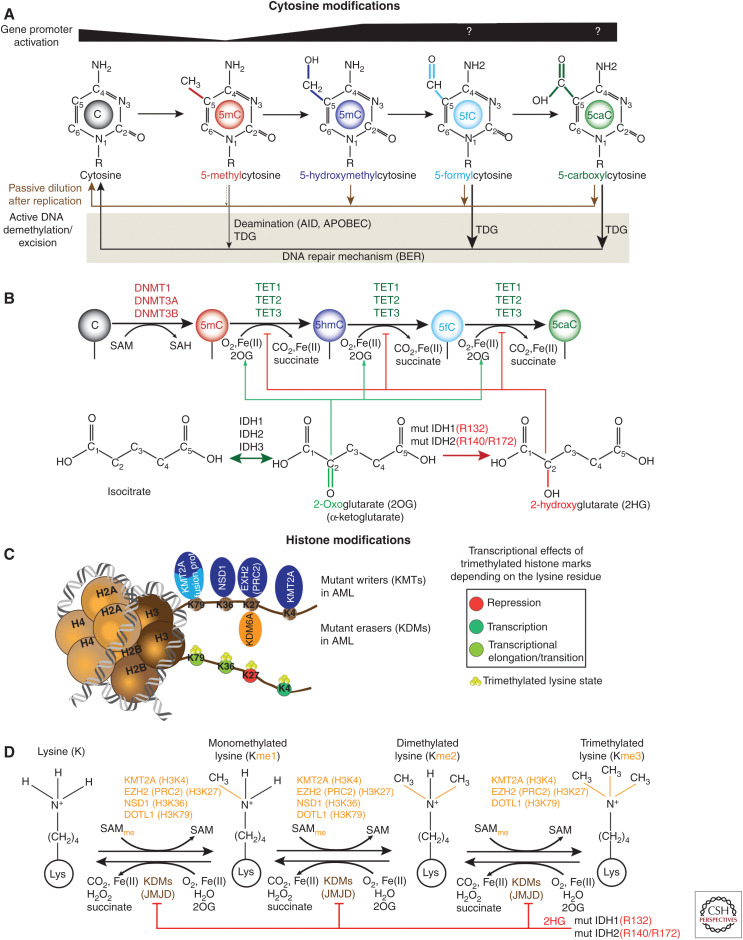
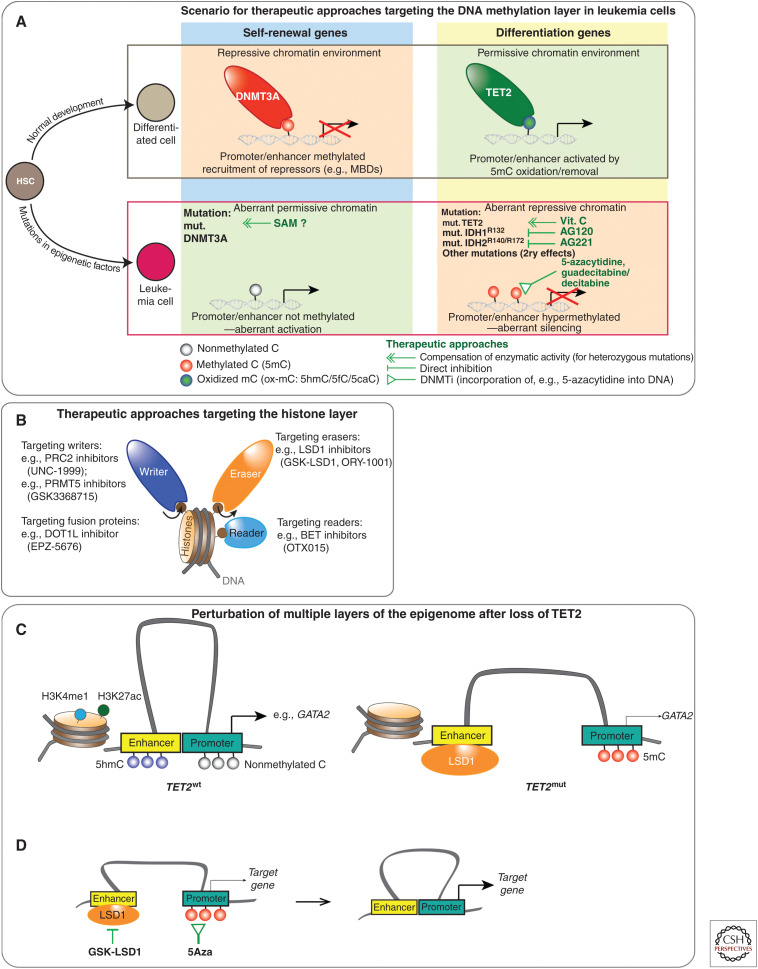
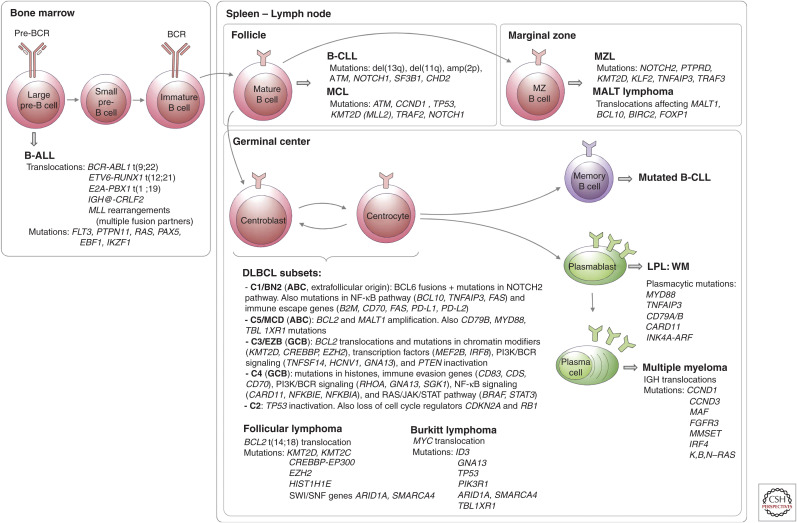
References
-
- Abbas S, Lugthart S, Kavelaars FG, Schelen A, Koenders J, Zeilemaker A, van Putten WJL, Rijneveld A, Löwenberg B, Valk PJM. 2010. Acquired mutations in the genes encoding IDH1 and IDH2 both are recurrent aberrations in acute myeloid leukemia (AML): prevalence and prognostic value. Blood 116: 2122–2126. 10.1182/blood-2009-11-250878 - DOI - PubMed
-
- Aldoss I, Yang D, Aribi A, Ali H, Sandhu K, Al Malki MM, Mei M, Salhotra A, Khaled S, Nakamura R, et al. 2018. Efficacy of the combination of venetoclax and hypomethylating agents in relapsed/refractory acute myeloid leukemia. Haematologica 103: e404–e407. 10.3324/haematol.2018.188094 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical