

Applying whole-genome sequencing in relation to phenotype and outcomes in siblings with cystic fibrosis
- PMID: 32014855
- PMCID: PMC6996517
- DOI: 10.1101/mcs.a004531
Applying whole-genome sequencing in relation to phenotype and outcomes in siblings with cystic fibrosis
Abstract
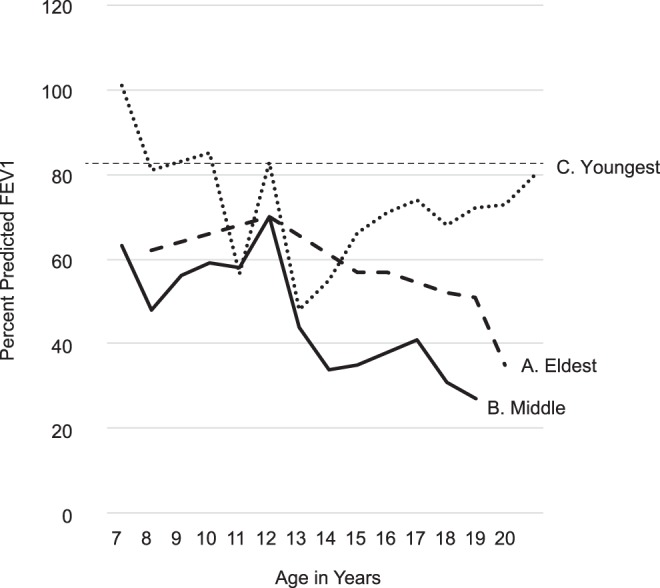
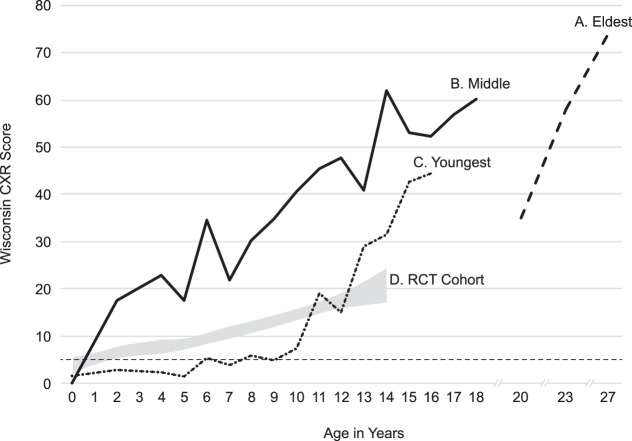
Variations in disease onset and/or severity have often been observed in siblings with cystic fibrosis (CF), despite the same CFTR genotype and environment. We postulated that genomic variation (modifier and/or pharmacogenomic variants) might explain these clinical discordances. From a cohort of patients included in the Wisconsin randomized clinical trial (RCT) of newborn screening (NBS) for CF, we identified two brothers who showed discordant lung disease courses as children, with one milder and the other more severe than average, and a third, eldest brother, who also has severe lung disease. Leukocytes were harvested as the source of DNA, and whole-genome sequencing (WGS) was performed. Variants were identified and analyzed using in-house-developed informatics tools. Lung disease onset and severity were quantitatively different between brothers during childhood. The youngest, less severely affected brother is homozygous for HFE p.H63D. He also has a very rare PLG p.D238N variant that may influence host-pathogen interaction during chronic lung infection. Other variants of interest were found differentially between the siblings. Pharmacogenomics findings were consistent with the middle, most severely affected brother having poor outcomes to common CF treatments. We conclude that genomic variation between siblings with CF is expected. Variable lung disease severity may be associated with differences acting as genetic modifiers and/or pharmacogenomic factors, but large cohort studies are needed to assess this hypothesis.
Keywords: bronchiectasis; chronic lung disease; diabetes mellitus; elevated sweat chloride; exocrine pancreatic insufficiency; meconium ileus; multiple bilateral pneumothoraces; recurrent Aspergillus infections; recurrent respiratory infections.
© 2020 Wilk et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Berge M, Guillemain R, Trégouet DA, Amrein C, Boussaud V, Chevalier P, Lillo-Lelouet A, Le Beller C, Laurent-Puig P, Beaune PH, et al. 2011. Effect of cytochrome P450 2C19 genotype on voriconazole exposure in cystic fibrosis lung transplant patients. Eur J Clin Pharmacol 67: 253–260. 10.1007/s00228-010-0914-2 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous