

Activation of ASC Inflammasome Driven by Toll-Like Receptor 4 Contributes to Host Immunity against Rickettsial Infection
- PMID: 32014896
- PMCID: PMC7093143
- DOI: 10.1128/IAI.00886-19
Activation of ASC Inflammasome Driven by Toll-Like Receptor 4 Contributes to Host Immunity against Rickettsial Infection
Abstract
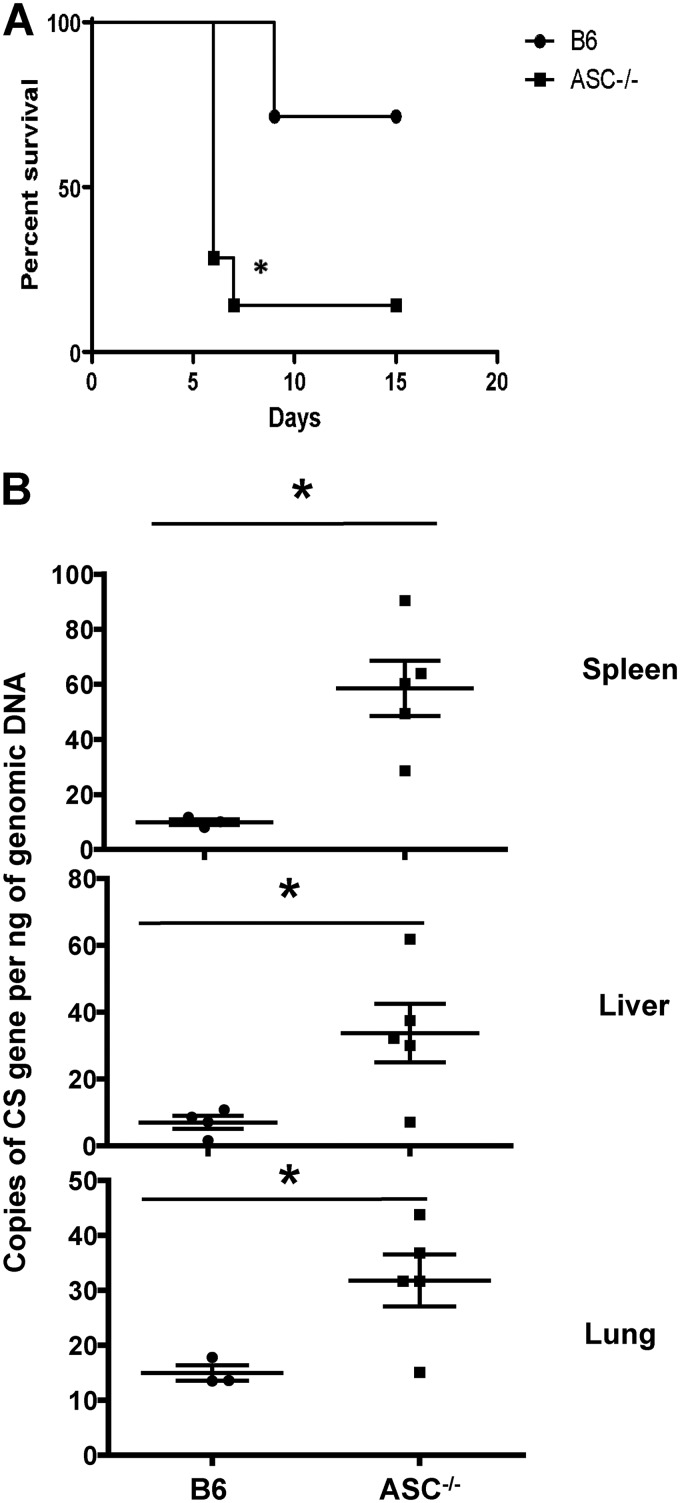
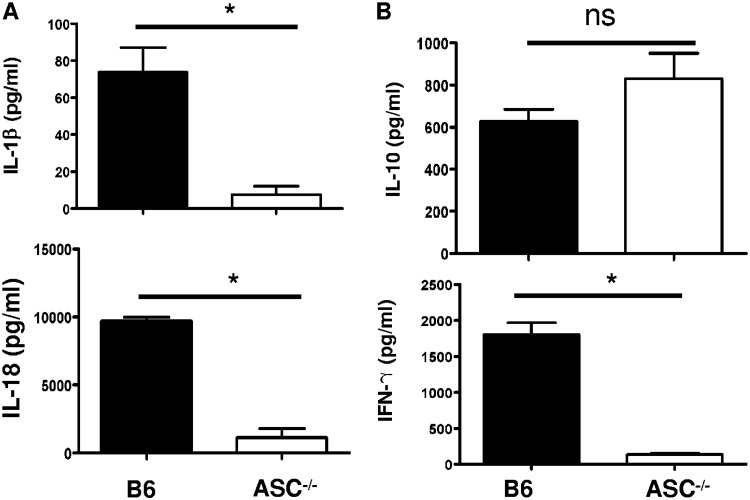
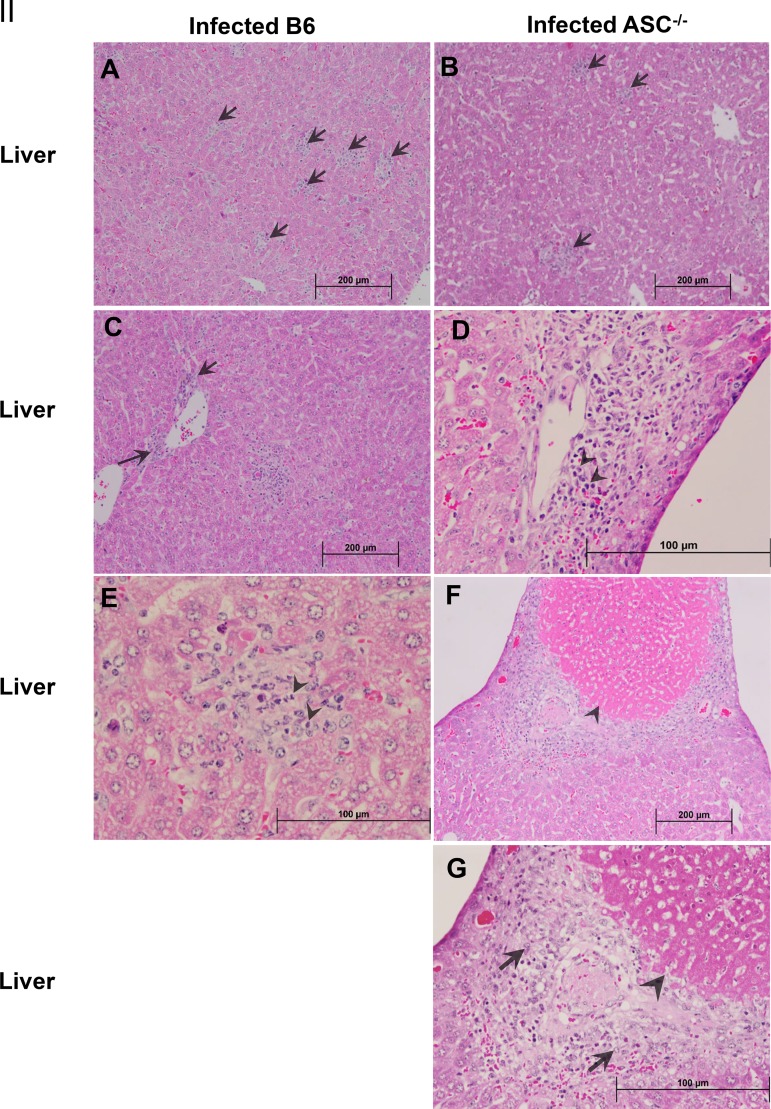
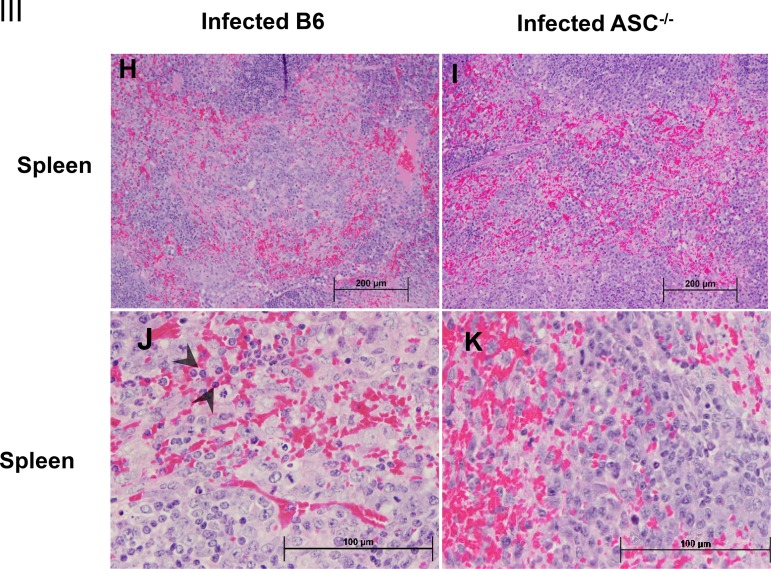
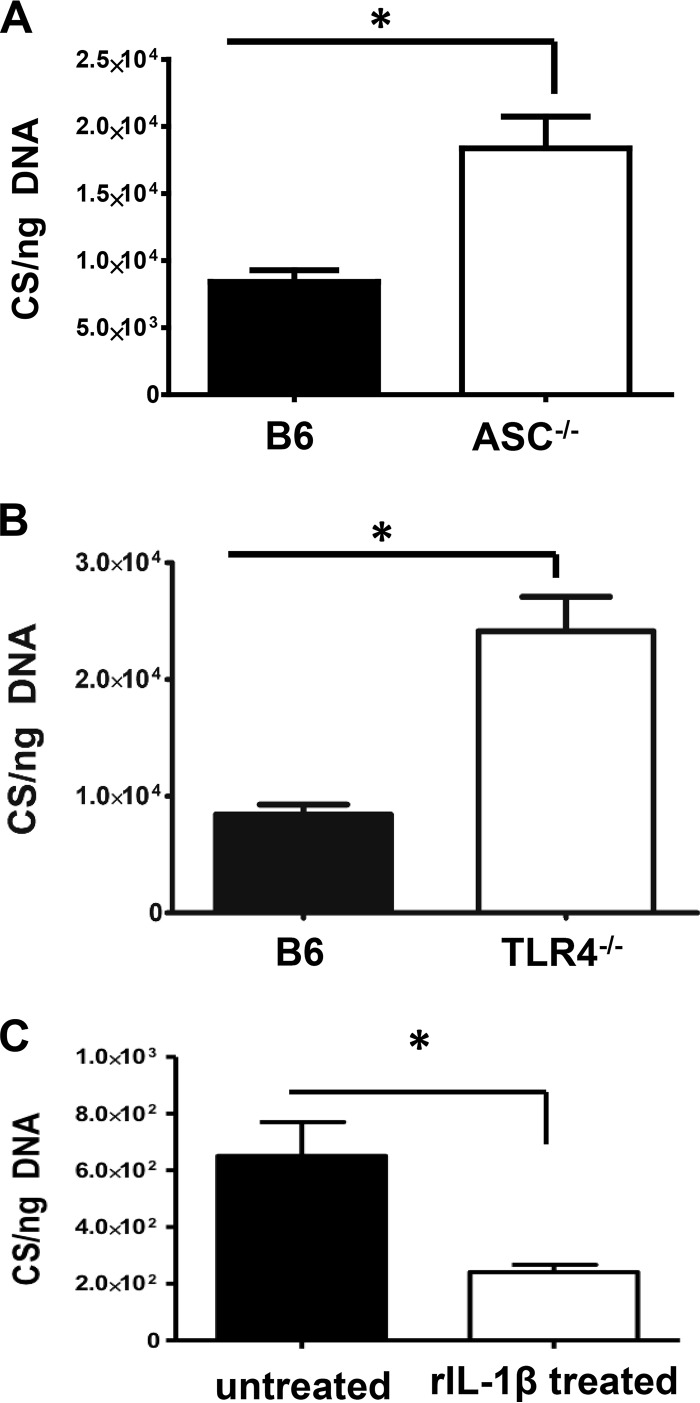
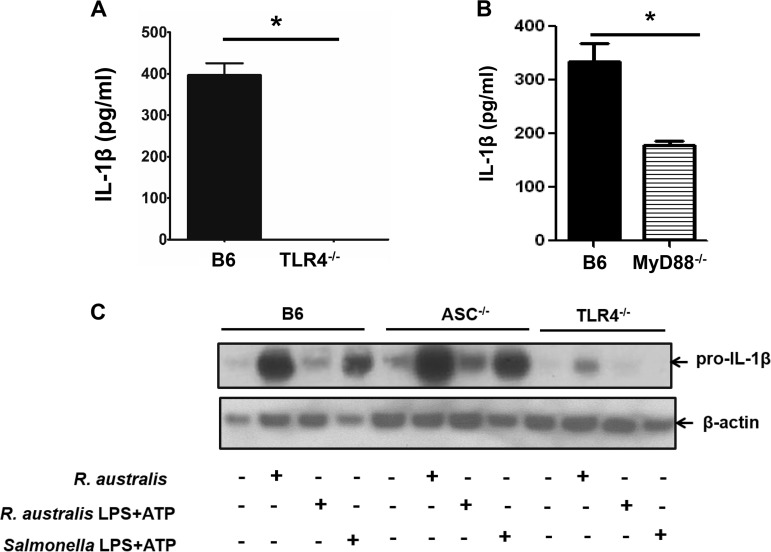
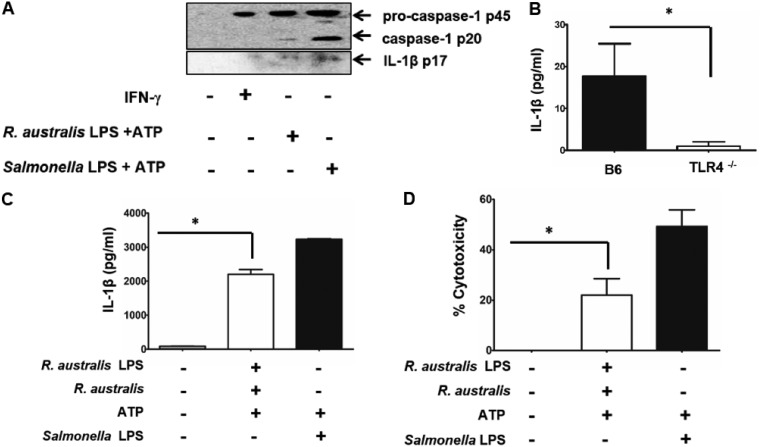
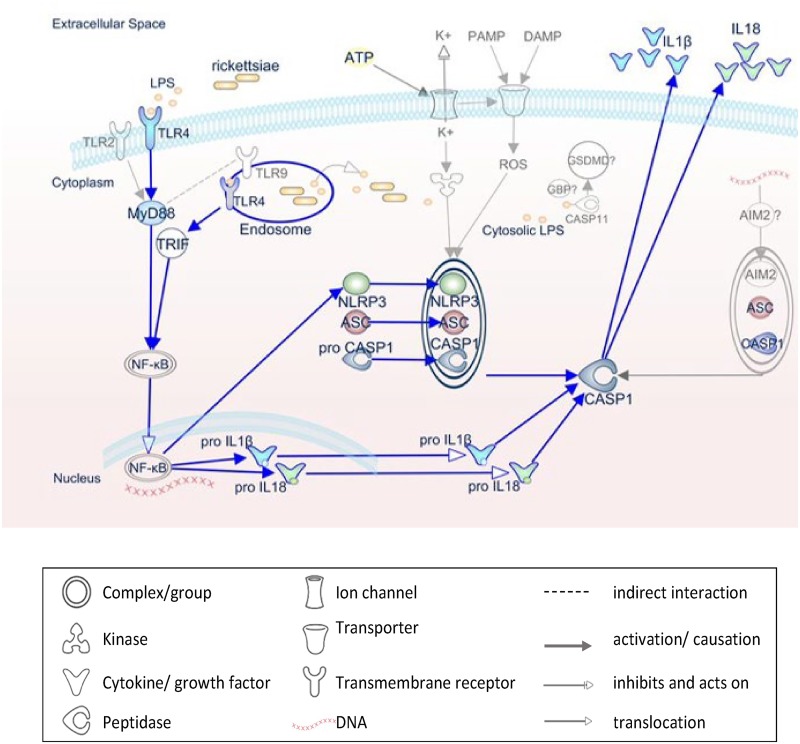
Rickettsiae are cytosolically replicating, obligately intracellular bacteria causing human infections worldwide with potentially fatal outcomes. We previously showed that Rickettsia australis activates ASC inflammasome in macrophages. In the present study, host susceptibility of ASC inflammasome-deficient mice to R. australis was significantly greater than that of C57BL/6 (B6) controls and was accompanied by increased rickettsial loads in various organs. Impaired host control of R. australis in vivo in ASC-/- mice was associated with dramatically reduced levels of interleukin 1β (IL-1β), IL-18, and gamma interferon (IFN-γ) in sera. The intracellular concentrations of R. australis in bone marrow-derived macrophages (BMMs) of TLR4-/- and ASC-/- mice were significantly greater than those in BMMs of B6 controls, highlighting the important role of inflammasome and these molecules in controlling rickettsiae in macrophages. Compared to B6 BMMs, TLR4-/- BMMs failed to secrete a significant level of IL-1β and had reduced expression levels of pro-IL-1β in response to infection with R. australis, suggesting that rickettsiae activate ASC inflammasome via a Toll-like receptor 4 (TLR4)-dependent mechanism. Further mechanistic studies suggest that the lipopolysaccharide (LPS) purified from R. australis together with ATP stimulation led to cleavage of pro-caspase-1 and pro-IL-1β, resulting in TLR4-dependent secretion of IL-1β. Taken together, these observations indicate that activation of ASC inflammasome, most likely driven by interaction of TLR4 with rickettsial LPS, contributes to host protective immunity against R. australis These findings provide key insights into defining the interactions of rickettsiae with the host innate immune system.
Keywords: ASC; LPS; TLR4; inflammasome; rickettsiae.
Copyright © 2020 American Society for Microbiology.
Figures

References
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
