

Identification of cancer driver genes based on nucleotide context
- PMID: 32015527
- PMCID: PMC7031046
- DOI: 10.1038/s41588-019-0572-y
Identification of cancer driver genes based on nucleotide context
Abstract
Cancer genomes contain large numbers of somatic mutations but few of these mutations drive tumor development. Current approaches either identify driver genes on the basis of mutational recurrence or approximate the functional consequences of nonsynonymous mutations by using bioinformatic scores. Passenger mutations are enriched in characteristic nucleotide contexts, whereas driver mutations occur in functional positions, which are not necessarily surrounded by a particular nucleotide context. We observed that mutations in contexts that deviate from the characteristic contexts around passenger mutations provide a signal in favor of driver genes. We therefore developed a method that combines this feature with the signals traditionally used for driver-gene identification. We applied our method to whole-exome sequencing data from 11,873 tumor-normal pairs and identified 460 driver genes that clustered into 21 cancer-related pathways. Our study provides a resource of driver genes across 28 tumor types with additional driver genes identified according to mutations in unusual nucleotide contexts.
Figures







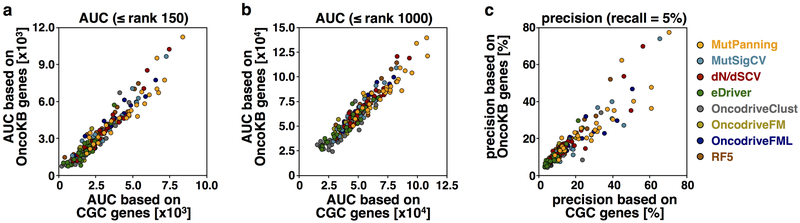
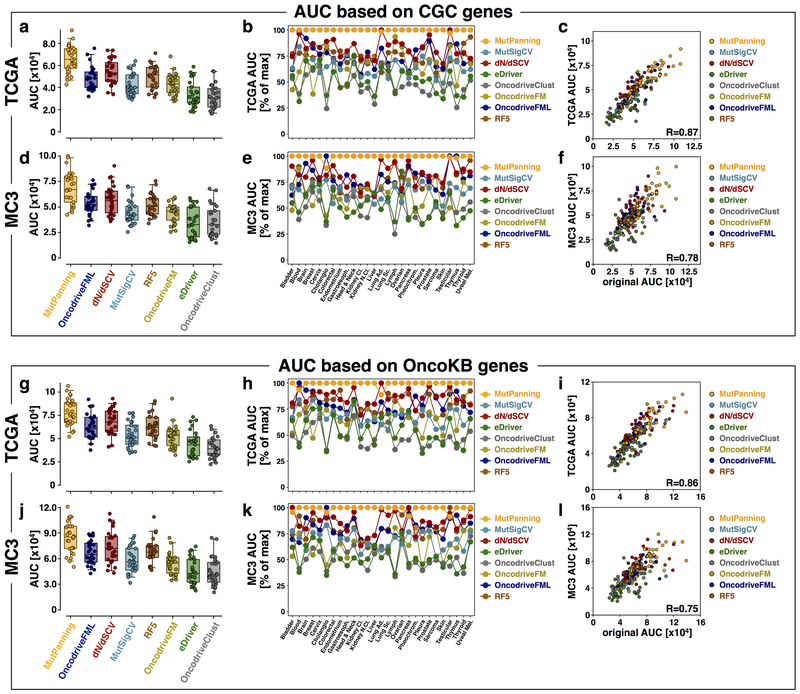



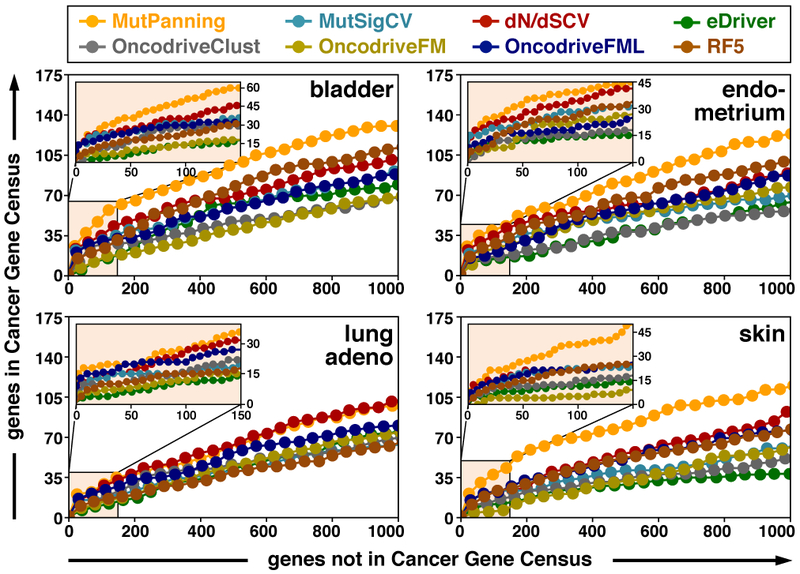

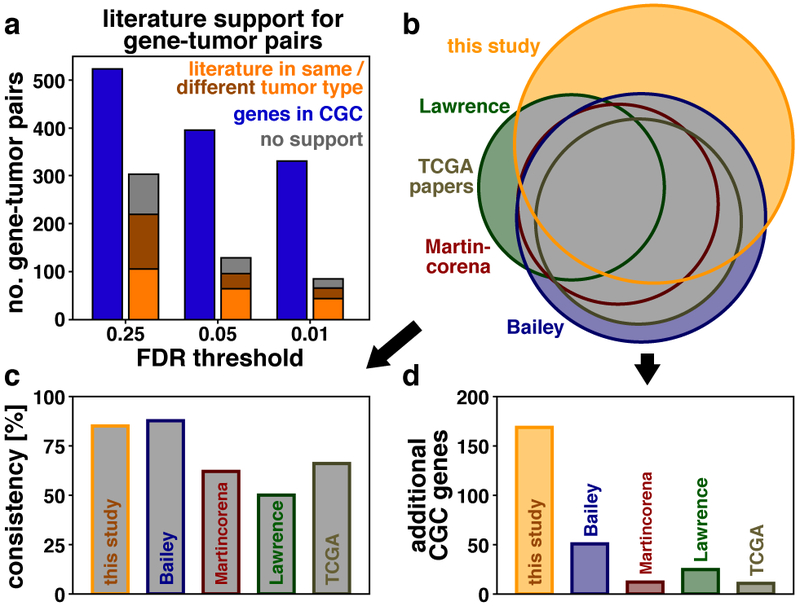

References
References (Online Methods)
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
