

Interplay between the tyrosine kinases Chk and Csk and phosphatase PTPRJ is critical for regulating platelets in mice
- PMID: 32016283
- PMCID: PMC7291955
- DOI: 10.1182/blood.2019002848
Interplay between the tyrosine kinases Chk and Csk and phosphatase PTPRJ is critical for regulating platelets in mice
Abstract
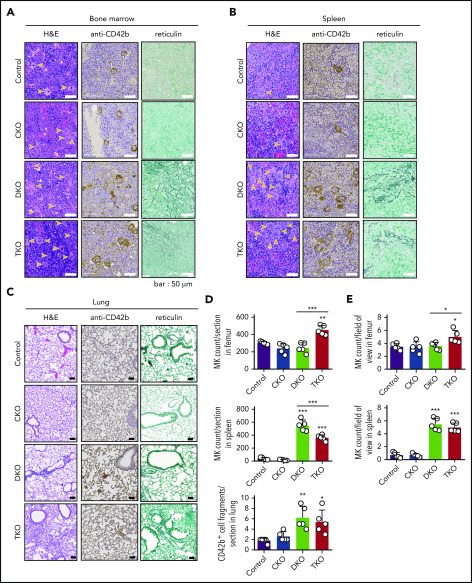
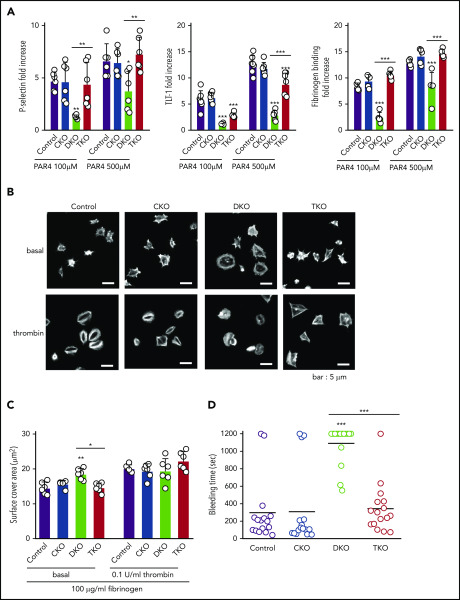
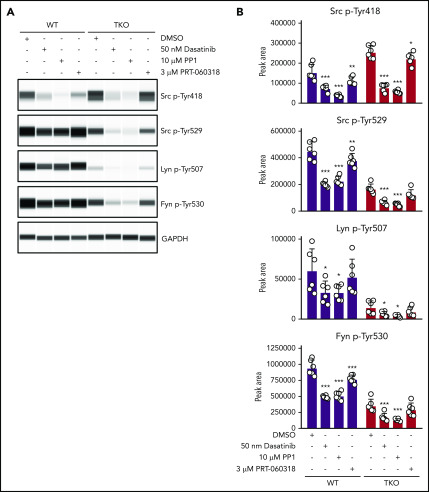
The Src family kinases (SFKs) Src, Lyn, and Fyn are essential for platelet activation and also involved in megakaryocyte (MK) development and platelet production. Platelet SFKs are inhibited by C-terminal Src kinase (Csk), which phosphorylates a conserved tyrosine in their C-terminal tail, and are activated by the receptor-type tyrosine phosphatase PTPRJ (CD148, DEP-1), which dephosphorylates the same residue. Deletion of Csk and PTPRJ in the MK lineage in mice results in increased SFK activity, but paradoxically hypoactive platelets resulting from negative feedback mechanisms, including upregulation of Csk homologous kinase (Chk) expression. Here, we investigate the role of Chk in platelets, functional redundancy with Csk, and the physiological consequences of ablating Chk, Csk, and PTPRJ in mice. Platelet count was normal in Chk knockout (KO) mice, reduced by 92% in Chk;Csk double KO (DKO) mice, and partially rescued in Chk;Csk;Ptprj triple KO (TKO) mice. Megakaryocyte numbers were significantly increased in both DKO and TKO mice. Phosphorylation of the inhibitory tyrosine of SFKs was almost completely abolished in DKO platelets, which was partially rescued in Src and Fyn in TKO platelets. This residual phosphorylation was abolished by Src inhibitors, revealing an unexpected mechanism in which SFKs autoinhibit their activity by phosphorylating their C-terminal tyrosine residues. We demonstrate that reduced inhibitory phosphorylation of SFKs leads to thrombocytopenia, with Csk being the dominant inhibitor in platelets and Chk having an auxiliary role. PTPRJ deletion in addition to Chk and Csk ameliorates the extent of thrombocytopenia, suggesting targeting it may have therapeutic benefits in such conditions.
© 2020 by The American Society of Hematology.
Conflict of interest statement
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Figures





References
-
- Mori J, Wang YJ, Ellison S, et al. . Dominant role of the protein-tyrosine phosphatase CD148 in regulating platelet activation relative to protein-tyrosine phosphatase-1B. Arterioscler Thromb Vasc Biol. 2012;32(12):2956-2965. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
