

Probing the mutational landscape of regulators of G protein signaling proteins in cancer
- PMID: 32019900
- PMCID: PMC7079226
- DOI: 10.1126/scisignal.aax8620
Probing the mutational landscape of regulators of G protein signaling proteins in cancer
Abstract
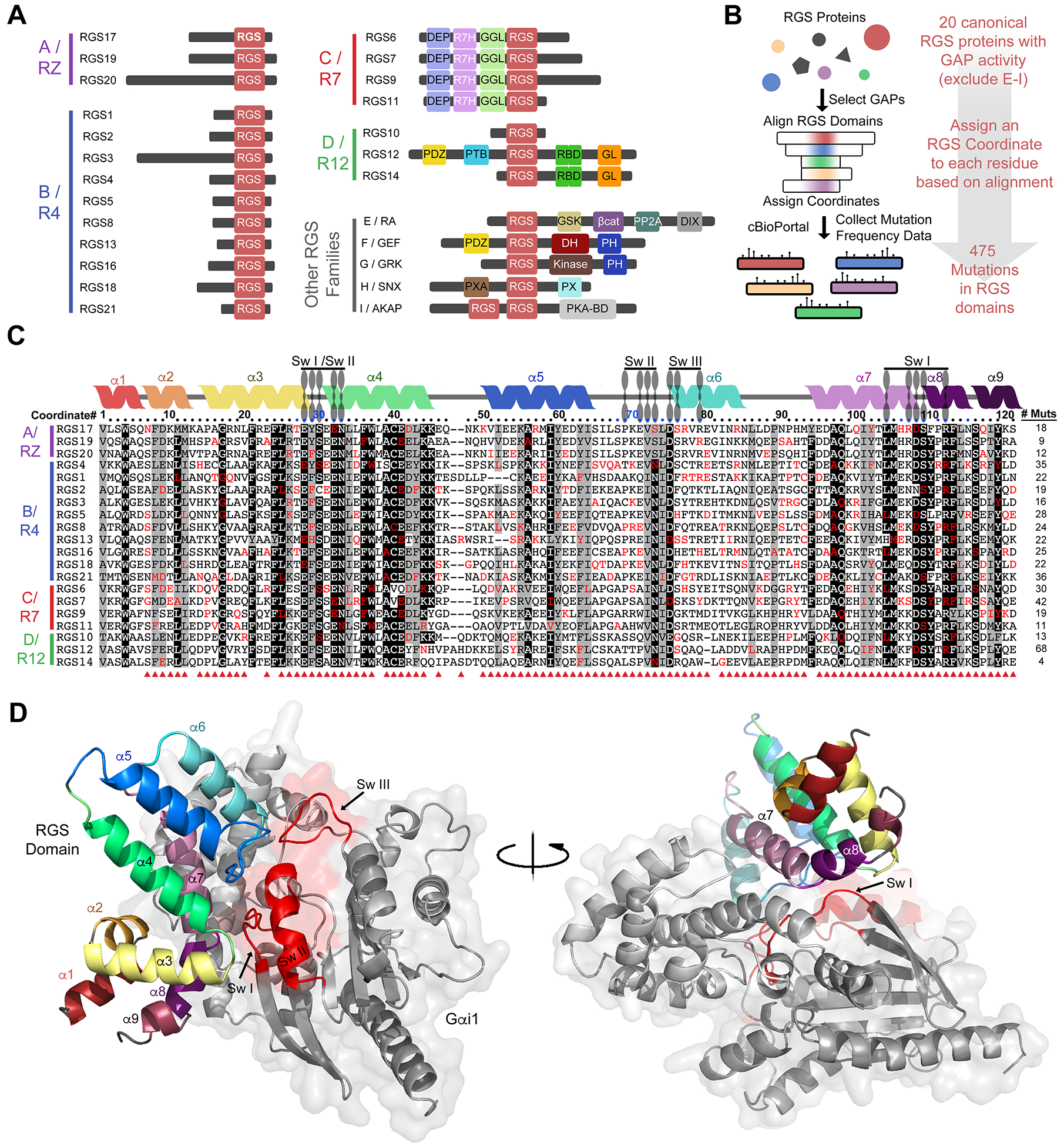
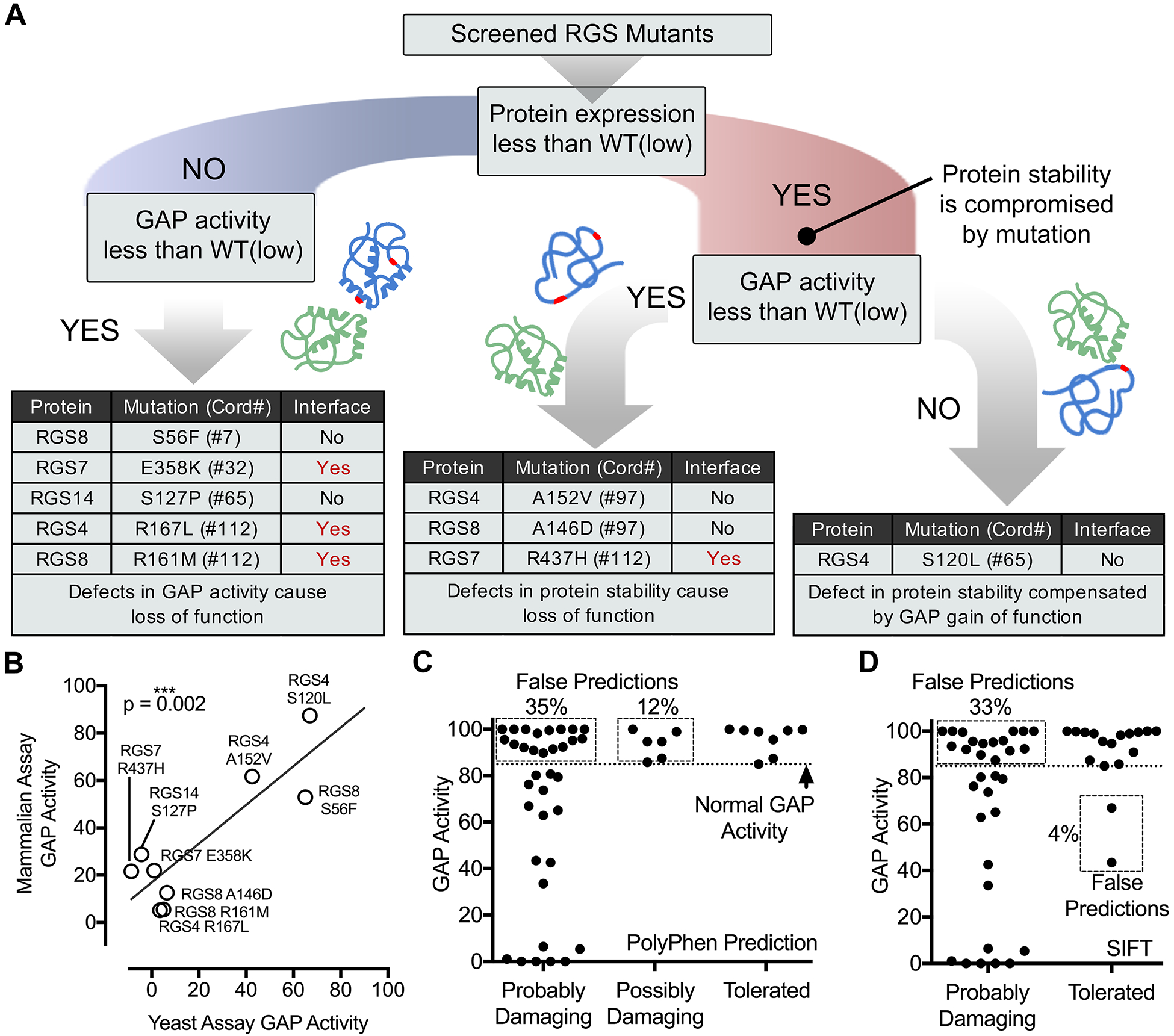
The advent of deep-sequencing techniques has revealed that mutations in G protein-coupled receptor (GPCR) signaling pathways in cancer are more prominent than was previously appreciated. An emergent theme is that cancer-associated mutations tend to cause enhanced GPCR pathway activation to favor oncogenicity. Regulators of G protein signaling (RGS) proteins are critical modulators of GPCR signaling that dampen the activity of heterotrimeric G proteins through their GTPase-accelerating protein (GAP) activity, which is conferred by a conserved domain dubbed the "RGS-box." Here, we developed an experimental pipeline to systematically assess the mutational landscape of RGS GAPs in cancer. A pan-cancer bioinformatics analysis of the 20 RGS domains with GAP activity revealed hundreds of low-frequency mutations spread throughout the conserved RGS domain structure with a slight enrichment at positions that interface with G proteins. We empirically tested multiple mutations representing all RGS GAP subfamilies and sampling both G protein interface and noninterface positions with a scalable, yeast-based assay. Last, a subset of mutants was validated using G protein activity biosensors in mammalian cells. Our findings reveal that a sizable fraction of RGS protein mutations leads to a loss of function through various mechanisms, including disruption of the G protein-binding interface, loss of protein stability, or allosteric effects on G protein coupling. Moreover, our results also validate a scalable pipeline for the rapid characterization of cancer-associated mutations in RGS proteins.
Copyright © 2020 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Conflict of interest statement
Figures





Similar articles
-
GTPase-activating proteins for heterotrimeric G proteins: regulators of G protein signaling (RGS) and RGS-like proteins.Annu Rev Biochem. 2000;69:795-827. doi: 10.1146/annurev.biochem.69.1.795. Annu Rev Biochem. 2000. PMID: 10966476 Review.
-
The GAPs, GEFs, and GDIs of heterotrimeric G-protein alpha subunits.Int J Biol Sci. 2005;1(2):51-66. doi: 10.7150/ijbs.1.51. Epub 2005 Apr 1. Int J Biol Sci. 2005. PMID: 15951850 Free PMC article. Review.
-
Regulators of G-protein signaling and their Gα substrates: promises and challenges in their use as drug discovery targets.Pharmacol Rev. 2011 Sep;63(3):728-49. doi: 10.1124/pr.110.003038. Epub 2011 Jul 7. Pharmacol Rev. 2011. PMID: 21737532 Free PMC article. Review.
-
Fluorescence-based assays for RGS box function.Methods Enzymol. 2004;389:56-71. doi: 10.1016/S0076-6879(04)89004-9. Methods Enzymol. 2004. PMID: 15313559 Review.
-
Interactions of the α-subunits of heterotrimeric G-proteins with GPCRs, effectors and RGS proteins: a critical review and analysis of interacting surfaces, conformational shifts, structural diversity and electrostatic potentials.J Struct Biol. 2013 Jun;182(3):209-18. doi: 10.1016/j.jsb.2013.03.004. Epub 2013 Mar 21. J Struct Biol. 2013. PMID: 23523730
Cited by
-
Heterotrimeric Gq proteins as therapeutic targets?J Biol Chem. 2020 Apr 17;295(16):5206-5215. doi: 10.1074/jbc.REV119.007061. Epub 2020 Mar 2. J Biol Chem. 2020. PMID: 32122969 Free PMC article. Review.
-
G protein regulation by RGS proteins in the pathophysiology of dilated cardiomyopathy.Am J Physiol Heart Circ Physiol. 2025 Feb 1;328(2):H348-H360. doi: 10.1152/ajpheart.00653.2024. Epub 2025 Jan 7. Am J Physiol Heart Circ Physiol. 2025. PMID: 39772618 Free PMC article. Review.
-
Fatty acid metabolism is related to the immune microenvironment changes of gastric cancer and RGS2 is a new tumor biomarker.Front Immunol. 2022 Dec 14;13:1065927. doi: 10.3389/fimmu.2022.1065927. eCollection 2022. Front Immunol. 2022. PMID: 36591293 Free PMC article.
-
Functional Assessment of Cancer-Linked Mutations in Sensitive Regions of Regulators of G Protein Signaling Predicted by Three-Dimensional Missense Tolerance Ratio Analysis.Mol Pharmacol. 2023 Jan;103(1):21-37. doi: 10.1124/molpharm.122.000614. Epub 2022 Nov 16. Mol Pharmacol. 2023. PMID: 36384958 Free PMC article.
-
Human genetic variants disrupt RGS14 nuclear shuttling and regulation of LTP in hippocampal neurons.J Biol Chem. 2021 Jan-Jun;296:100024. doi: 10.1074/jbc.RA120.016009. Epub 2020 Nov 22. J Biol Chem. 2021. PMID: 33410399 Free PMC article.
References
-
- Marx J, Nobel Prizes. Medicine: a signal award for discovering G proteins. Science (New York, N.Y 266, 368–369 (October 21, 1994). - PubMed
-
- Gilman AG, G proteins: transducers of receptor-generated signals. Annual review of biochemistry 56, 615–649 (1987). - PubMed
-
- Hopkins AL, Groom CR, The druggable genome. Nature reviews. Drug discovery 1, 727–730 (September, 2002). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
