

Transcriptional effects of copy number alterations in a large set of human cancers
- PMID: 32024838
- PMCID: PMC7002723
- DOI: 10.1038/s41467-020-14605-5
Transcriptional effects of copy number alterations in a large set of human cancers
Abstract
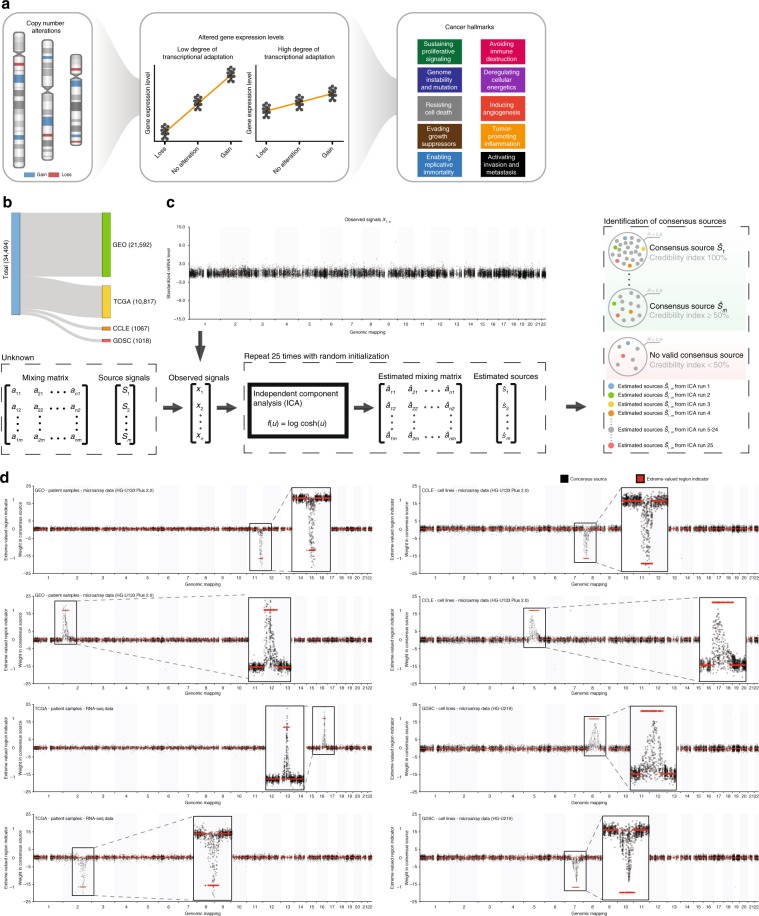
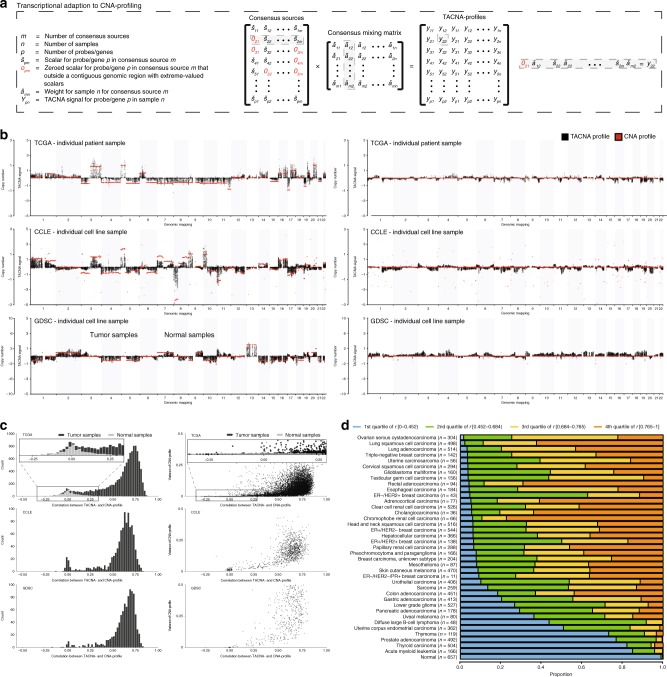
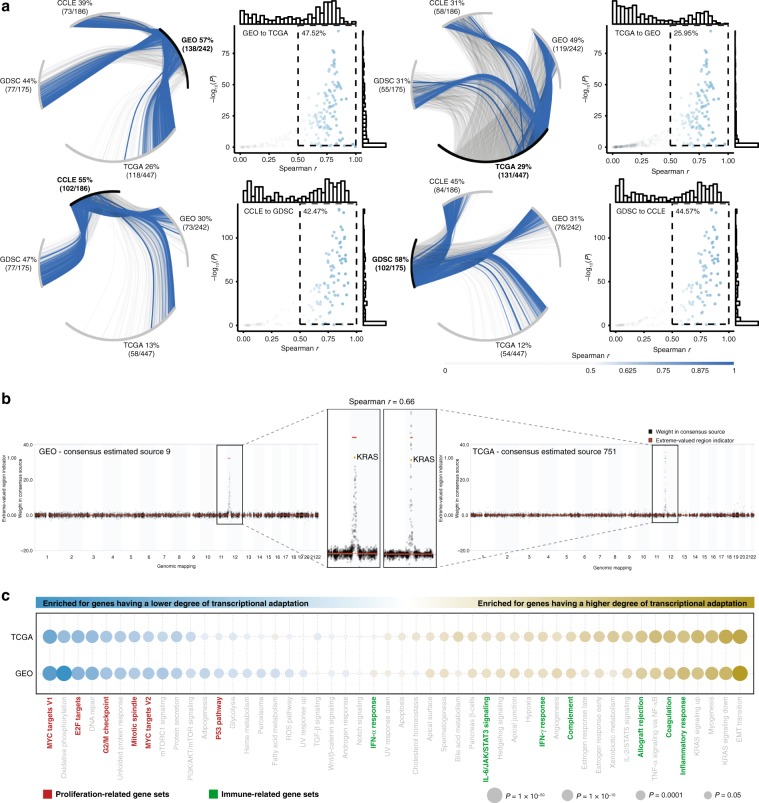
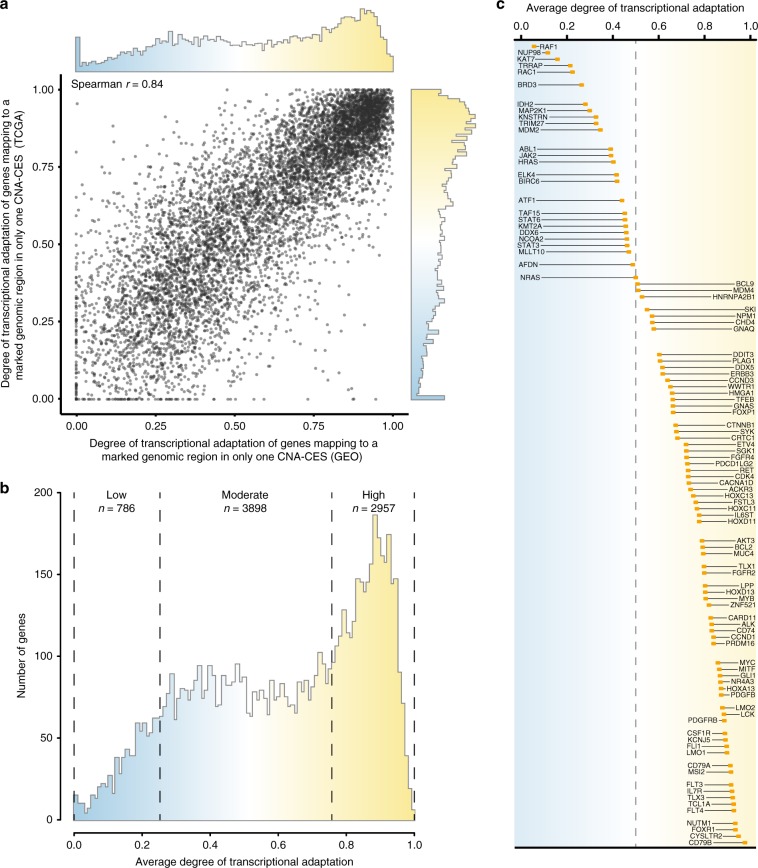
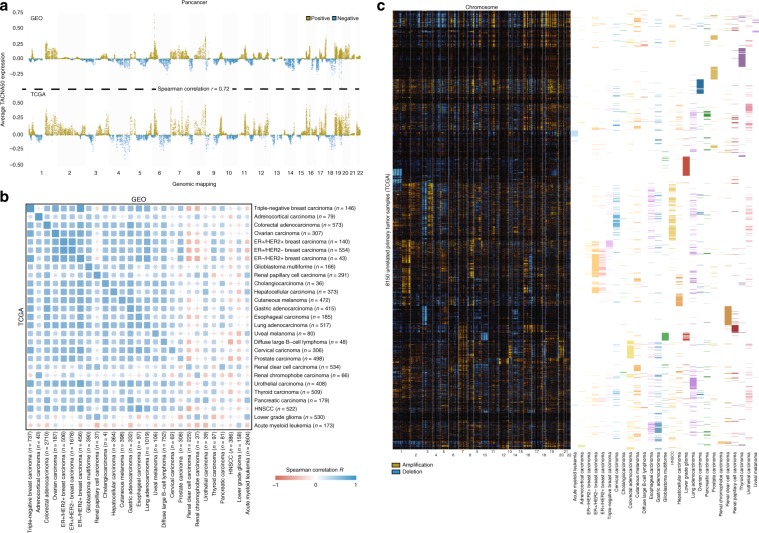
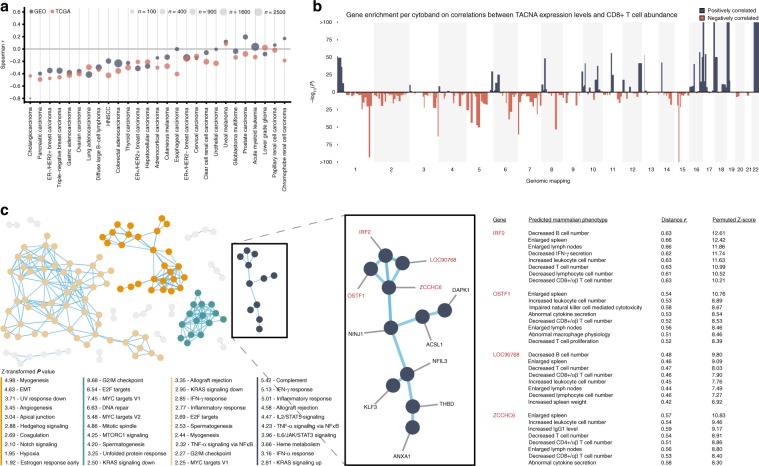
Copy number alterations (CNAs) can promote tumor progression by altering gene expression levels. Due to transcriptional adaptive mechanisms, however, CNAs do not always translate proportionally into altered expression levels. By reanalyzing >34,000 gene expression profiles, we reveal the degree of transcriptional adaptation to CNAs in a genome-wide fashion, which strongly associate with distinct biological processes. We then develop a platform-independent method-transcriptional adaptation to CNA profiling (TACNA profiling)-that extracts the transcriptional effects of CNAs from gene expression profiles without requiring paired CNA profiles. By applying TACNA profiling to >28,000 patient-derived tumor samples we define the landscape of transcriptional effects of CNAs. The utility of this landscape is demonstrated by the identification of four genes that are predicted to be involved in tumor immune evasion when transcriptionally affected by CNAs. In conclusion, we provide a novel tool to gain insight into how CNAs drive tumor behavior via altered expression levels.
Conflict of interest statement
E.G.E.d.V. reports institutional financial support for advisory board/consultancy from Sanofi, Daiichi, Sankyo, NSABP, Pfizer and Merck, and institutional financial support for clinical trials or contracted research from Amgen, Genentech, Roche, AstraZeneca, Synthon, Nordic Nanovector, G1 Therapeutics, Bayer, Chugai Pharma, CytomX Therapeutics and Radius Health, all unrelated to the submitted work. All other authors declare no competing interests.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
