

Inferring structural variant cancer cell fraction
- PMID: 32024845
- PMCID: PMC7002525
- DOI: 10.1038/s41467-020-14351-8
Inferring structural variant cancer cell fraction
Erratum in
-
Author Correction: Inferring structural variant cancer cell fraction.Nat Commun. 2022 Dec 8;13(1):7568. doi: 10.1038/s41467-022-32338-5. Nat Commun. 2022. PMID: 36481724 Free PMC article. No abstract available.
Abstract
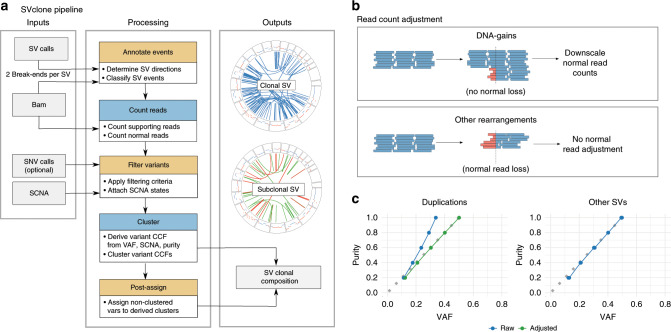
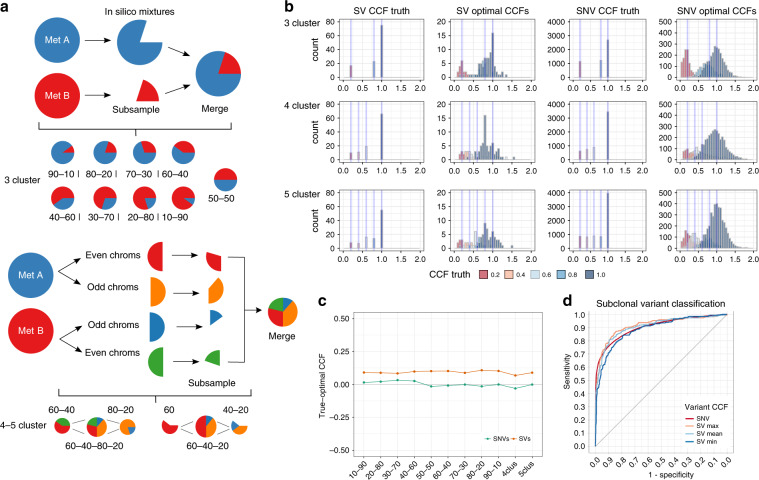
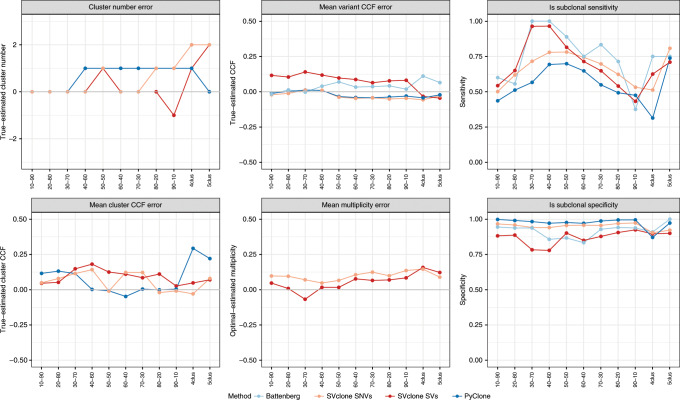
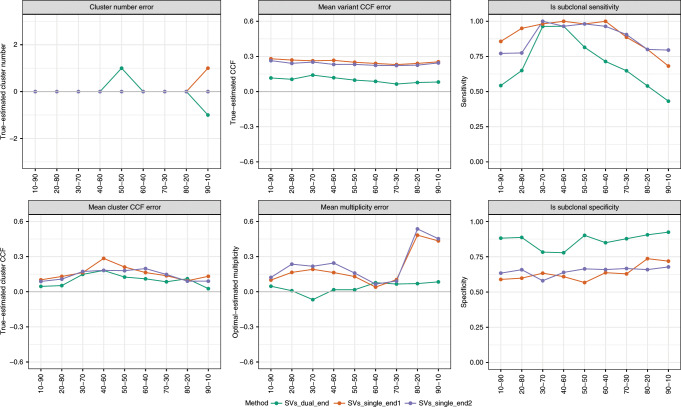
We present SVclone, a computational method for inferring the cancer cell fraction of structural variant (SV) breakpoints from whole-genome sequencing data. SVclone accurately determines the variant allele frequencies of both SV breakends, then simultaneously estimates the cancer cell fraction and SV copy number. We assess performance using in silico mixtures of real samples, at known proportions, created from two clonal metastases from the same patient. We find that SVclone's performance is comparable to single-nucleotide variant-based methods, despite having an order of magnitude fewer data points. As part of the Pan-Cancer Analysis of Whole Genomes (PCAWG) consortium, which aggregated whole-genome sequencing data from 2658 cancers across 38 tumour types, we use SVclone to reveal a subset of liver, ovarian and pancreatic cancers with subclonally enriched copy-number neutral rearrangements that show decreased overall survival. SVclone enables improved characterisation of SV intra-tumour heterogeneity.
Conflict of interest statement
R.B. owns equity in Ampressa Therapeutics. G.G. receives research funds from IBM and Pharmacyclics and is an inventor on patent applications related to MuTect, ABSOLUTE, MutSig, MSMuTect and POLYSOLVER. I.L. is a consultant for PACT Pharma. B.J.R. is a consultant at and has ownership interest (including stock, patents, etc.) in Medley Genomics. All the other authors have no competing interests.
Figures
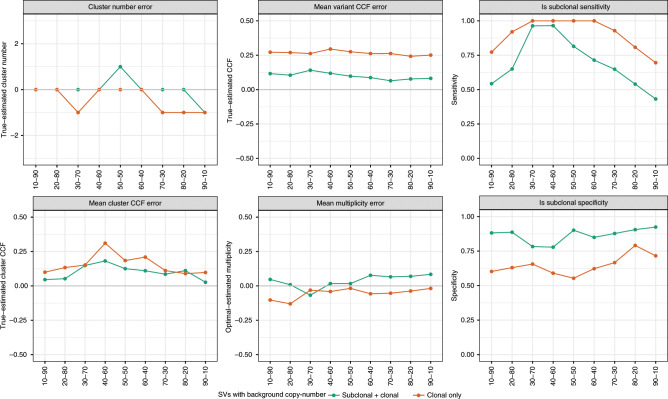
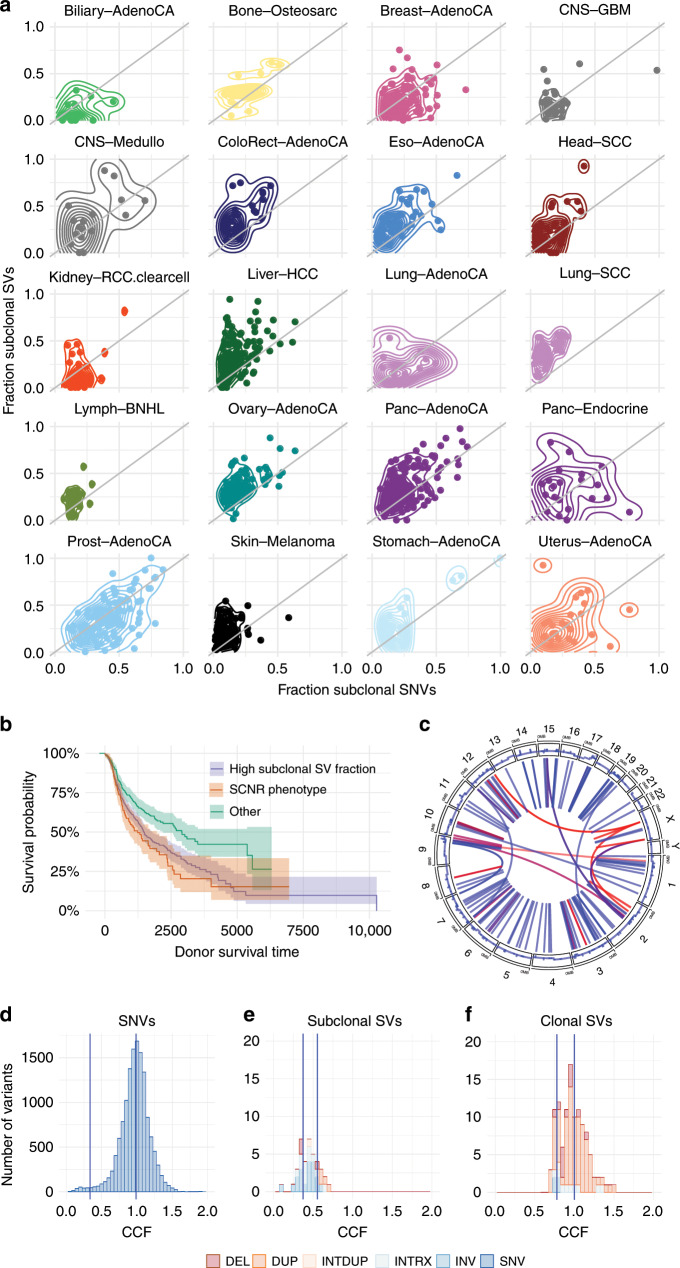
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
