

Integrative pathway enrichment analysis of multivariate omics data
- PMID: 32024846
- PMCID: PMC7002665
- DOI: 10.1038/s41467-019-13983-9
Integrative pathway enrichment analysis of multivariate omics data
Erratum in
-
Author Correction: Integrative pathway enrichment analysis of multivariate omics data.Nat Commun. 2022 Dec 8;13(1):7570. doi: 10.1038/s41467-022-32342-9. Nat Commun. 2022. PMID: 36481658 Free PMC article. No abstract available.
Abstract
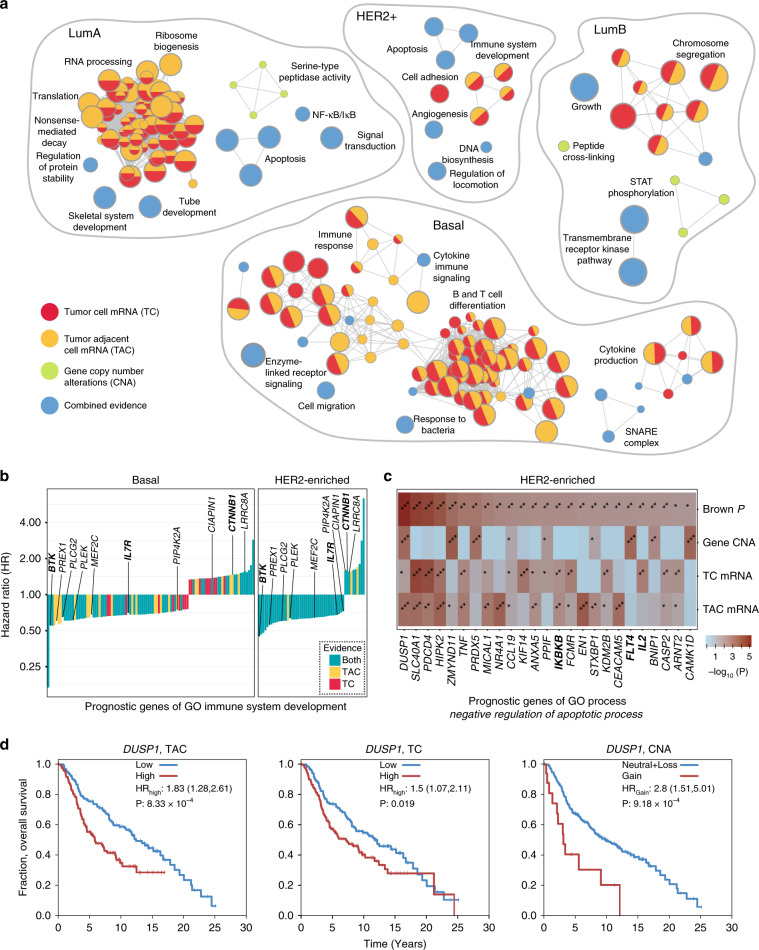
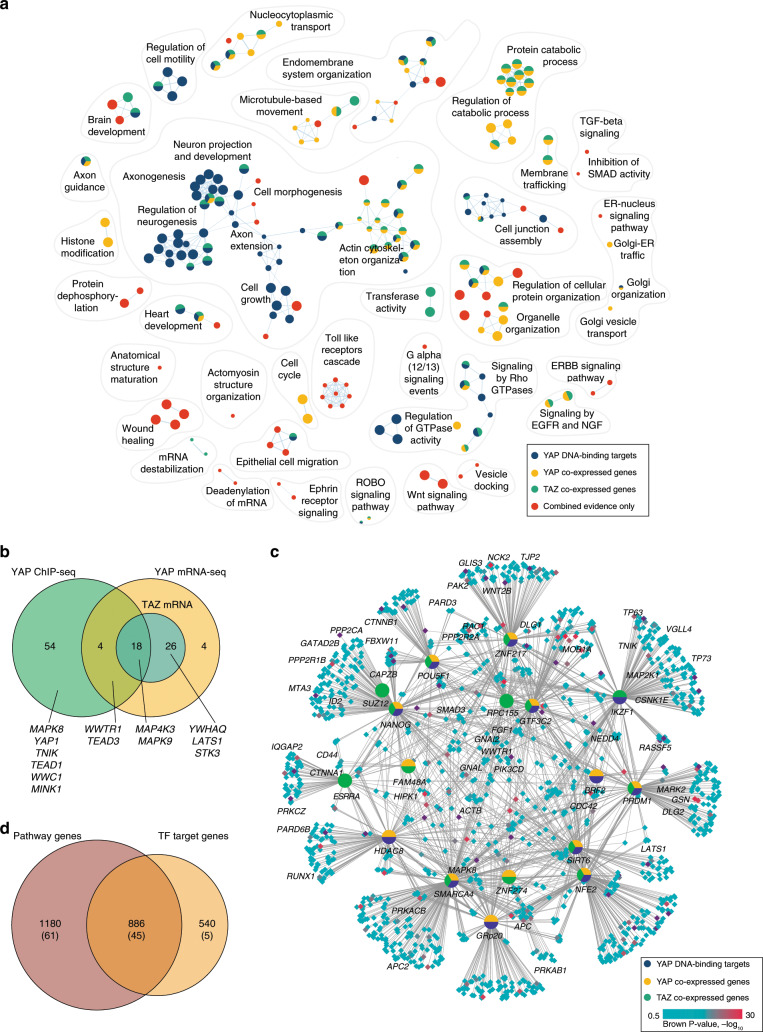
Multi-omics datasets represent distinct aspects of the central dogma of molecular biology. Such high-dimensional molecular profiles pose challenges to data interpretation and hypothesis generation. ActivePathways is an integrative method that discovers significantly enriched pathways across multiple datasets using statistical data fusion, rationalizes contributing evidence and highlights associated genes. As part of the ICGC/TCGA Pan-Cancer Analysis of Whole Genomes (PCAWG) Consortium, which aggregated whole genome sequencing data from 2658 cancers across 38 tumor types, we integrated genes with coding and non-coding mutations and revealed frequently mutated pathways and additional cancer genes with infrequent mutations. We also analyzed prognostic molecular pathways by integrating genomic and transcriptomic features of 1780 breast cancers and highlighted associations with immune response and anti-apoptotic signaling. Integration of ChIP-seq and RNA-seq data for master regulators of the Hippo pathway across normal human tissues identified processes of tissue regeneration and stem cell regulation. ActivePathways is a versatile method that improves systems-level understanding of cellular organization in health and disease through integration of multiple molecular datasets and pathway annotations.
Conflict of interest statement
The authors declare no competing interests.
Figures



References
-
- The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature10.1038/s41586-020-1969-6 (2020).
