

Pathway and network analysis of more than 2500 whole cancer genomes
- PMID: 32024854
- PMCID: PMC7002574
- DOI: 10.1038/s41467-020-14367-0
Pathway and network analysis of more than 2500 whole cancer genomes
Erratum in
-
Author Correction: Pathway and network analysis of more than 2500 whole cancer genomes.Nat Commun. 2022 Dec 8;13(1):7566. doi: 10.1038/s41467-022-32334-9. Nat Commun. 2022. PMID: 36481610 Free PMC article. No abstract available.
Abstract
The catalog of cancer driver mutations in protein-coding genes has greatly expanded in the past decade. However, non-coding cancer driver mutations are less well-characterized and only a handful of recurrent non-coding mutations, most notably TERT promoter mutations, have been reported. Here, as part of the ICGC/TCGA Pan-Cancer Analysis of Whole Genomes (PCAWG) Consortium, which aggregated whole genome sequencing data from 2658 cancer across 38 tumor types, we perform multi-faceted pathway and network analyses of non-coding mutations across 2583 whole cancer genomes from 27 tumor types compiled by the ICGC/TCGA PCAWG project that was motivated by the success of pathway and network analyses in prioritizing rare mutations in protein-coding genes. While few non-coding genomic elements are recurrently mutated in this cohort, we identify 93 genes harboring non-coding mutations that cluster into several modules of interacting proteins. Among these are promoter mutations associated with reduced mRNA expression in TP53, TLE4, and TCF4. We find that biological processes had variable proportions of coding and non-coding mutations, with chromatin remodeling and proliferation pathways altered primarily by coding mutations, while developmental pathways, including Wnt and Notch, altered by both coding and non-coding mutations. RNA splicing is primarily altered by non-coding mutations in this cohort, and samples containing non-coding mutations in well-known RNA splicing factors exhibit similar gene expression signatures as samples with coding mutations in these genes. These analyses contribute a new repertoire of possible cancer genes and mechanisms that are altered by non-coding mutations and offer insights into additional cancer vulnerabilities that can be investigated for potential therapeutic treatments.
Conflict of interest statement
P.B. receives grant funding from Novartis from an unrelated project. R.B. owns equity in Ampressa Therapeutics. G.G. receives research funds from IBM and Pharmacyclics and is an inventor on patent applications related to MuTect, ABSOLUTE, MutSig, MSMuTect, and POLYSOLVER. B.J.R. is a consultant at and has an ownership interest (including stock, patents, etc.) in Medley Genomics. Remaining authors have no competing interests.
Figures
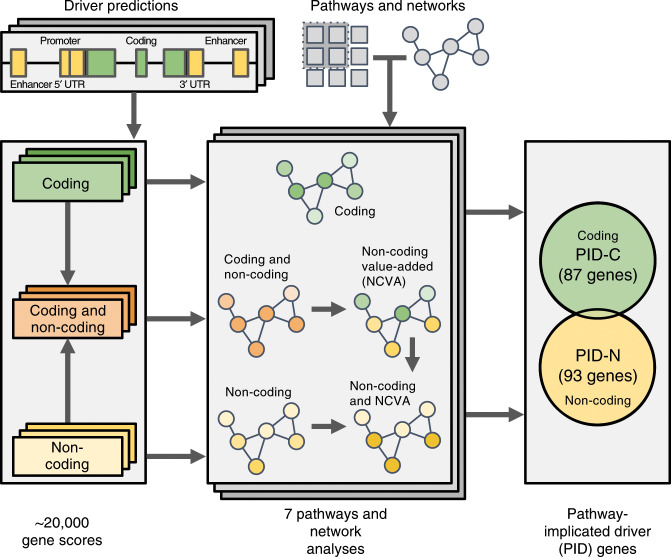
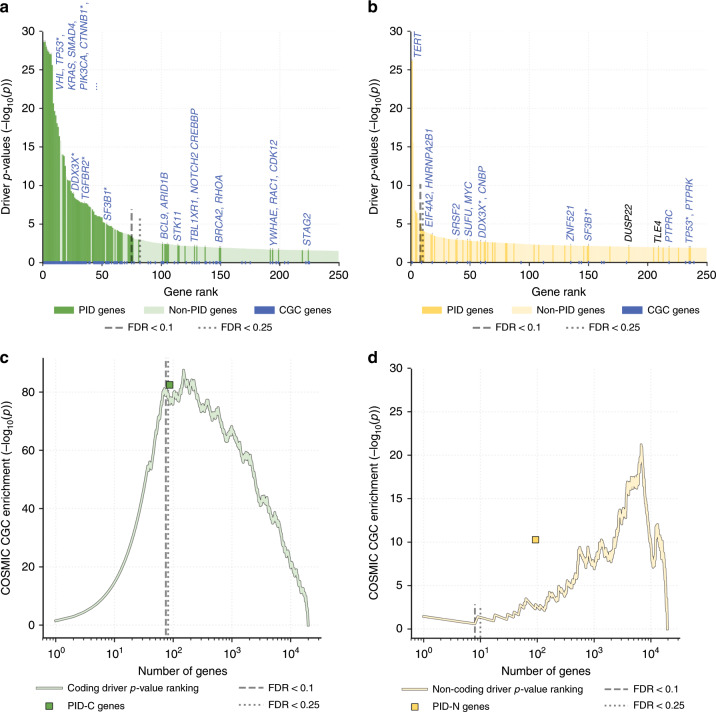


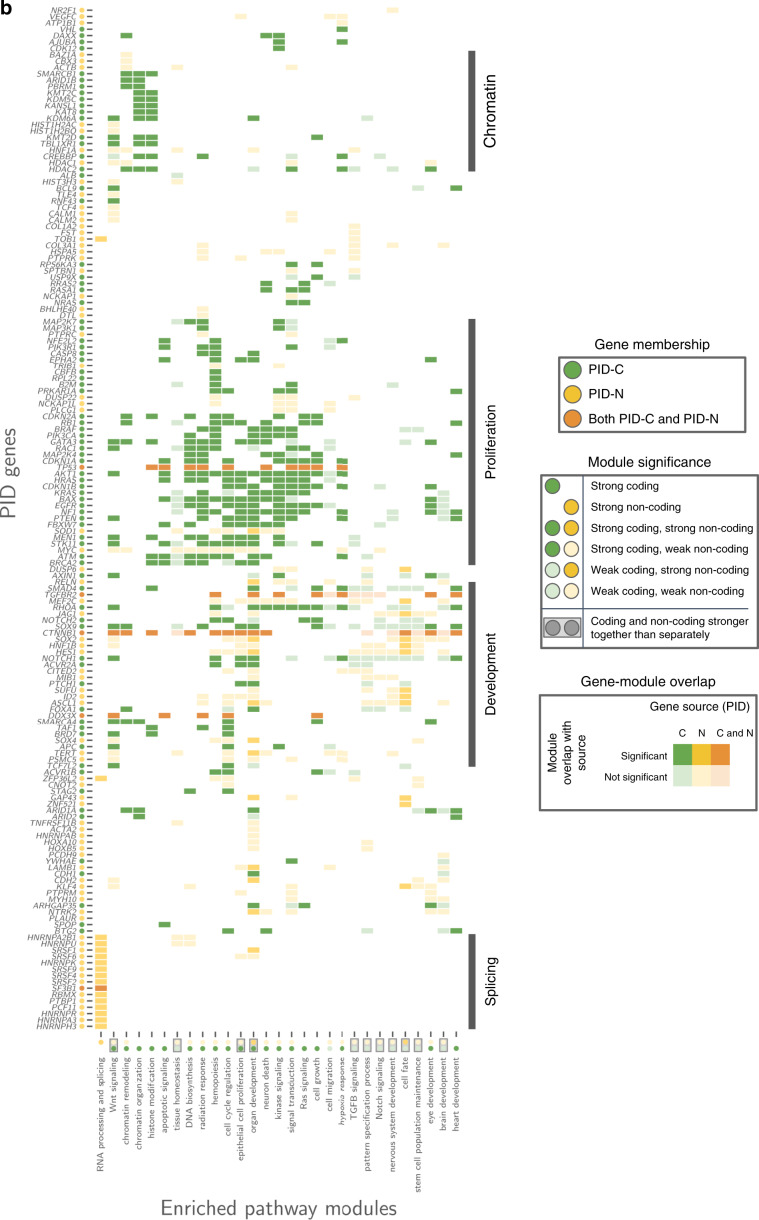
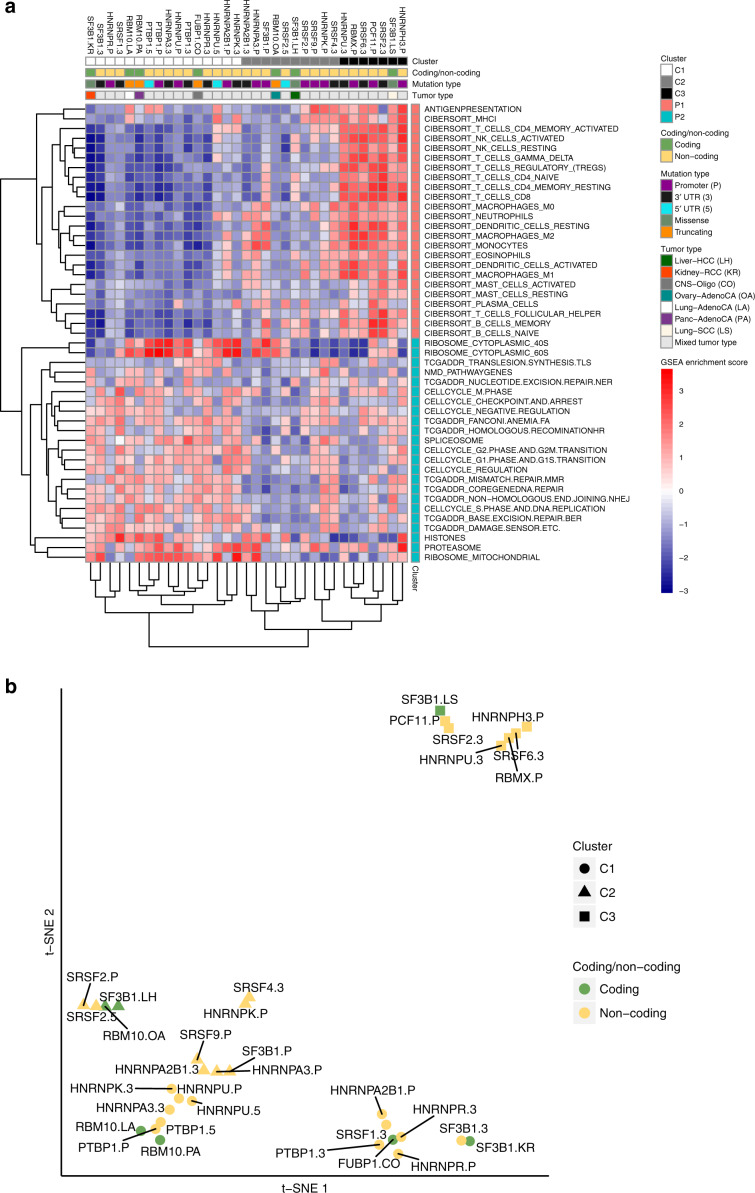
References
Publication types
MeSH terms
Grants and funding
- 23924/CRUK_/Cancer Research UK/United Kingdom
- U01 CA217875/CA/NCI NIH HHS/United States
- P30 CA008748/CA/NCI NIH HHS/United States
- R01 CA218668/CA/NCI NIH HHS/United States
- U24 CA143858/CA/NCI NIH HHS/United States
- T32 GM008313/GM/NIGMS NIH HHS/United States
- R01 CA180778/CA/NCI NIH HHS/United States
- U24 CA210990/CA/NCI NIH HHS/United States
- U24 CA211000/CA/NCI NIH HHS/United States
- P30 ES010126/ES/NIEHS NIH HHS/United States
- P30 CA016672/CA/NCI NIH HHS/United States
- R01 GM109031/GM/NIGMS NIH HHS/United States
- R01 HG007069/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
