

CD8+ T cell states in human cancer: insights from single-cell analysis
- PMID: 32024970
- PMCID: PMC7115982
- DOI: 10.1038/s41568-019-0235-4
CD8+ T cell states in human cancer: insights from single-cell analysis
Abstract
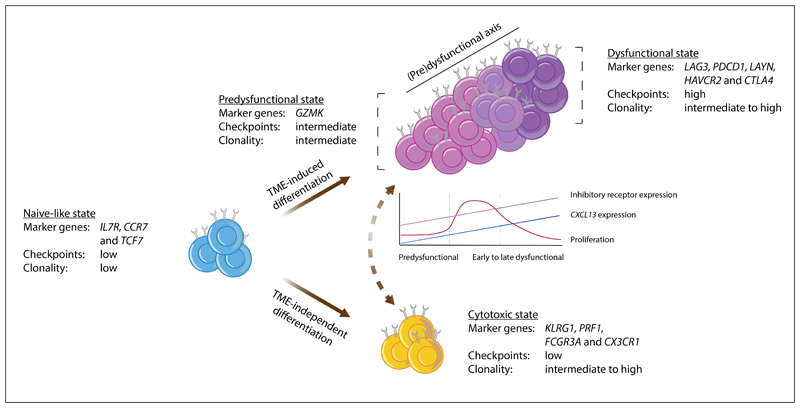
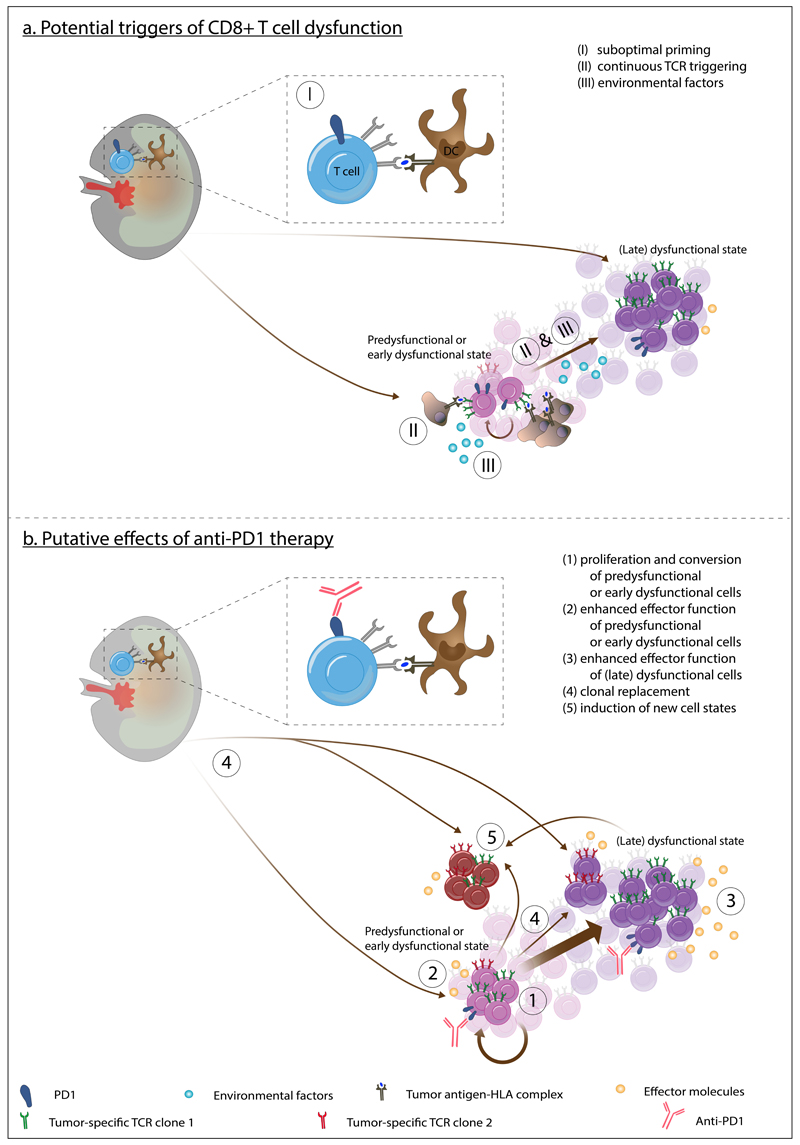
The T cell infiltrates that are formed in human cancers are a modifier of natural disease progression and also determine the probability of clinical response to cancer immunotherapies. Recent technological advances that allow the single-cell analysis of phenotypic and transcriptional states have revealed a vast heterogeneity of intratumoural T cell states, both within and between patients, and the observation of this heterogeneity makes it critical to understand the relationship between individual T cell states and therapy response. This Review covers our current knowledge of the T cell states that are present in human tumours and the role that different T cell populations have been hypothesized to play within the tumour microenvironment, with a particular focus on CD8+ T cells. The three key models that are discussed herein are as follows: (1) the dysfunction of T cells in human cancer is associated with a change in T cell functionality rather than inactivity; (2) antigen recognition in the tumour microenvironment is an important driver of T cell dysfunctionality and the presence of dysfunctional T cells can hence be used as a proxy for the presence of a tumour-reactive T cell compartment; (3) a less dysfunctional population of tumour-reactive T cells may be required to drive a durable response to T cell immune checkpoint blockade.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Clemente CG, et al. Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer. 1996;77:1303–1310. - PubMed
-
- Galon J, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science (80-. ) 2006;313:1960–1964. - PubMed
-
- Zhang L, et al. Intratumoral T Cells, Recurrence, and Survival in Epithelial Ovarian Cancer. N Engl J Med. 2003;348:203–213. - PubMed
-
- Fridman WH, Pages F, Sauts-Fridman C, Galon J. The immune contexture in human tumours: Impact on clinical outcome. Nature Reviews Cancer. 2012;12:298–306. - PubMed
Highlighted references
-
-
Azizi et al., Cell (2018): Single cell transcriptome analysis of immune cells in human breast cancer demonstrating that intratumoral T cells reside along a continuum that is driven by activation and terminal differentiation. In addition, cell state diversity was shown both between and within T cell clones.
-
-
-
Clarke et al., J. Exp. Med. (2019): Single cell transcriptome analysis of T cells in human lung cancer describing a PD1 and TIM3 expressing TRM cell subset that contains a highly proliferative subset and is enriched in lesions of patients responding to anti-PD1 therapy.
-
-
-
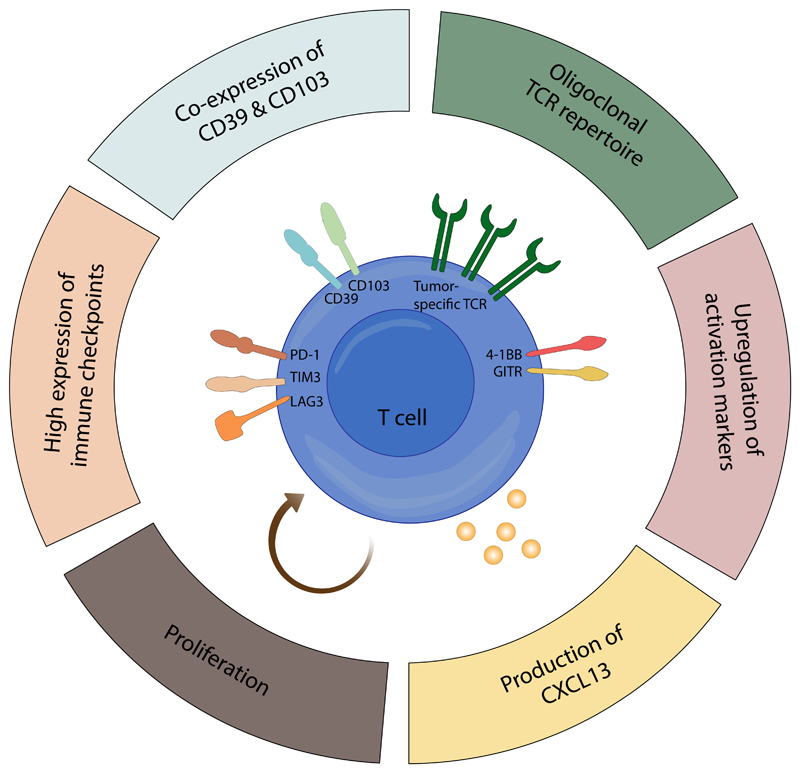
Duhen et al., Nature Communications (2018): This study identifies CD103 and CD39 as markers of tumor-reactive T cells across multiple human cancer types.
-
-
-
Guo et al., Nature Medicine (2018): Single cell transcriptome analysis of T cells in NSCLC, adjacent normal tissue, and blood showing the distribution of T cell states in these tissues and their relatedness based on TCR sharing between cell states in and outside the tumor. Moreover, this study provides evidence for dysfunctionality as a gradual state.
-
-
-
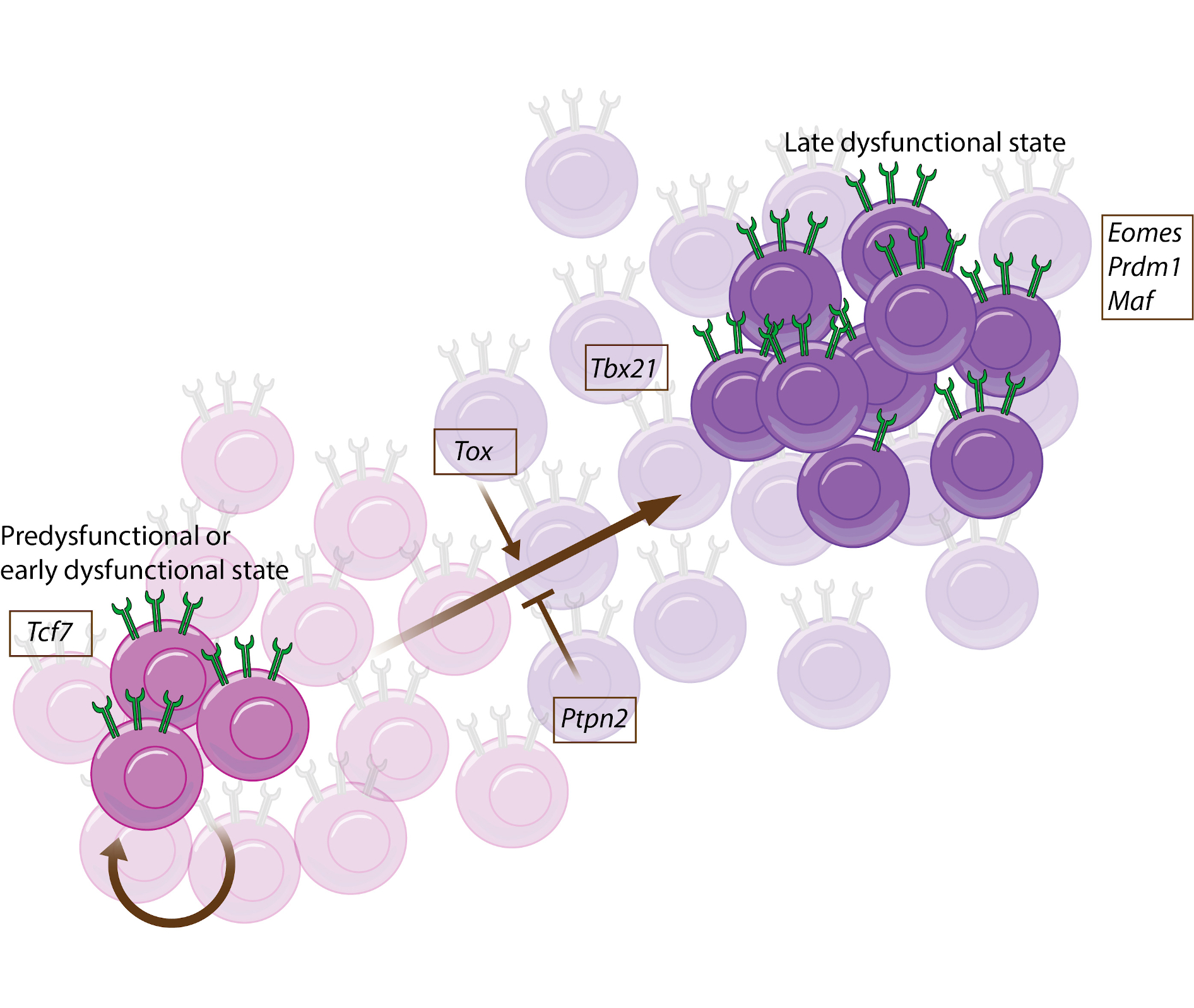
Kurtulus et al., Immunity (2019): A mouse study that describes a PD1 low subset of T cells that respond to ICB and identifies Tcf7 expression to be required for response to anti-PD1 and anti-TIM3 combination therapy.
-
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
