Cancer LncRNA Census reveals evidence for deep functional conservation of long noncoding RNAs in tumorigenesis
- PMID: 32024996
- PMCID: PMC7002399
- DOI: 10.1038/s42003-019-0741-7
Cancer LncRNA Census reveals evidence for deep functional conservation of long noncoding RNAs in tumorigenesis
Erratum in
-
Author Correction: Cancer LncRNA Census reveals evidence for deep functional conservation of long noncoding RNAs in tumorigenesis.Commun Biol. 2022 Dec 8;5(1):1347. doi: 10.1038/s42003-022-03769-z. Commun Biol. 2022. PMID: 36482178 Free PMC article. No abstract available.
Abstract
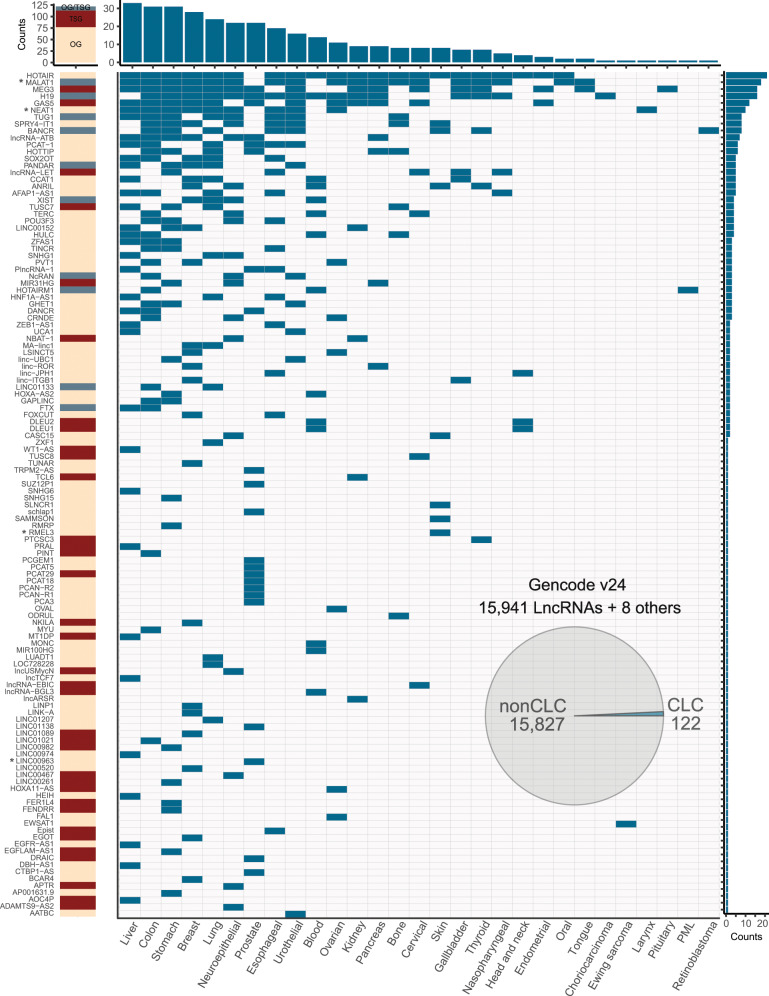
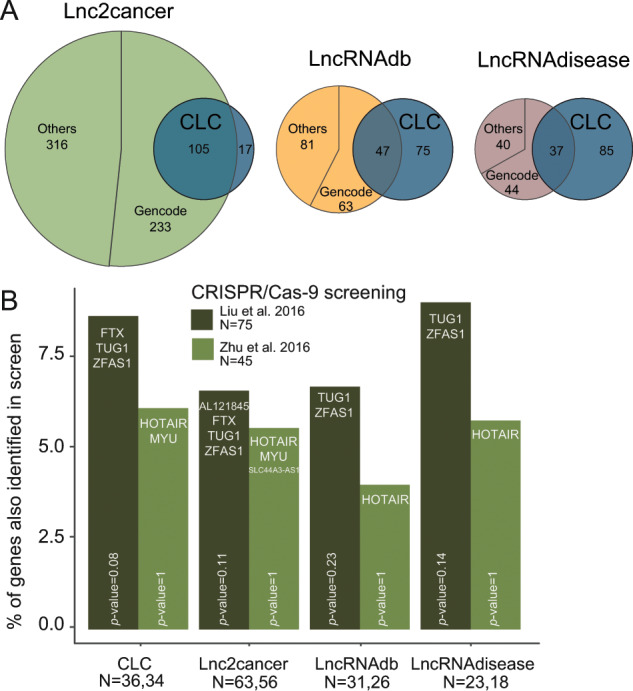
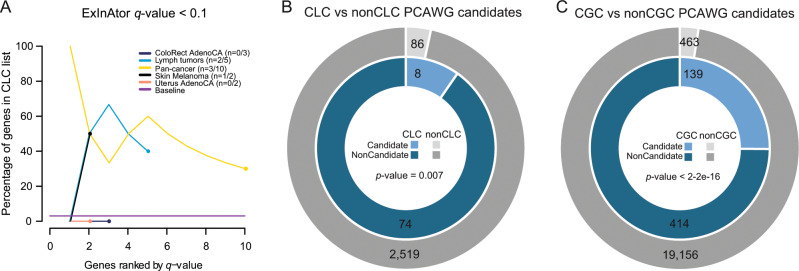
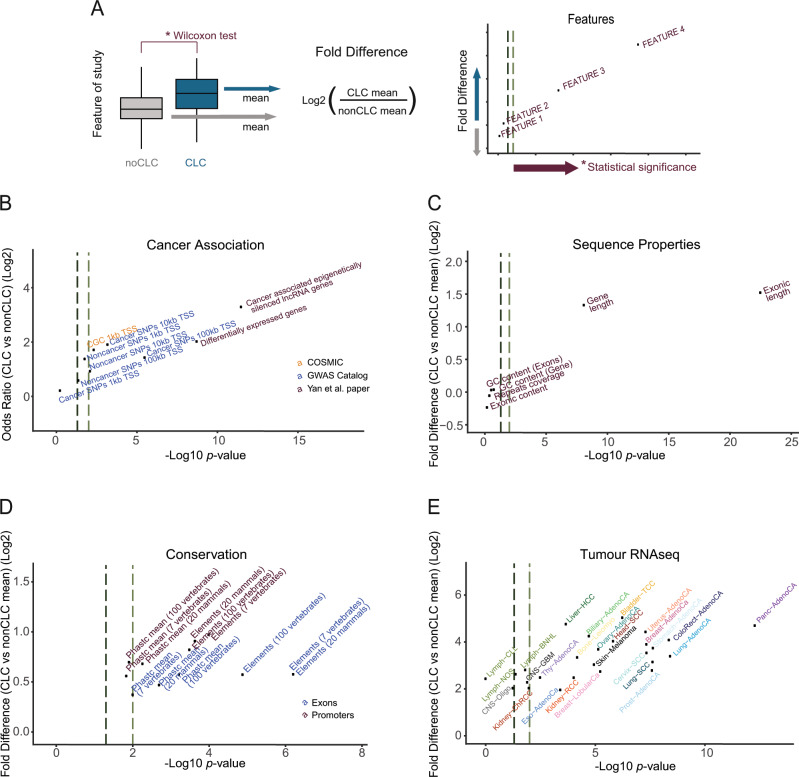
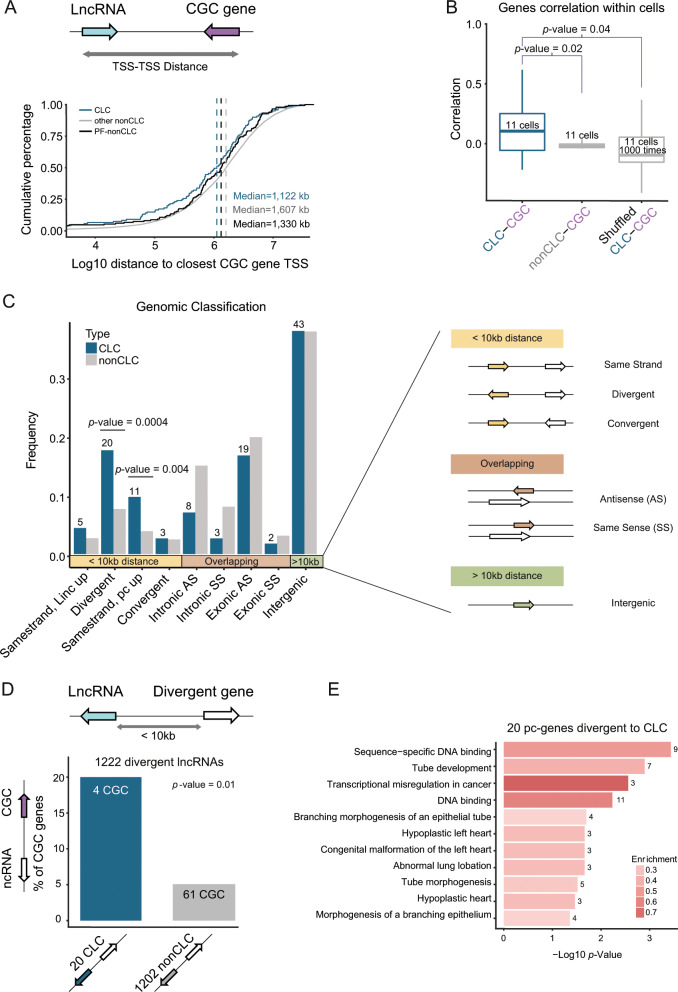
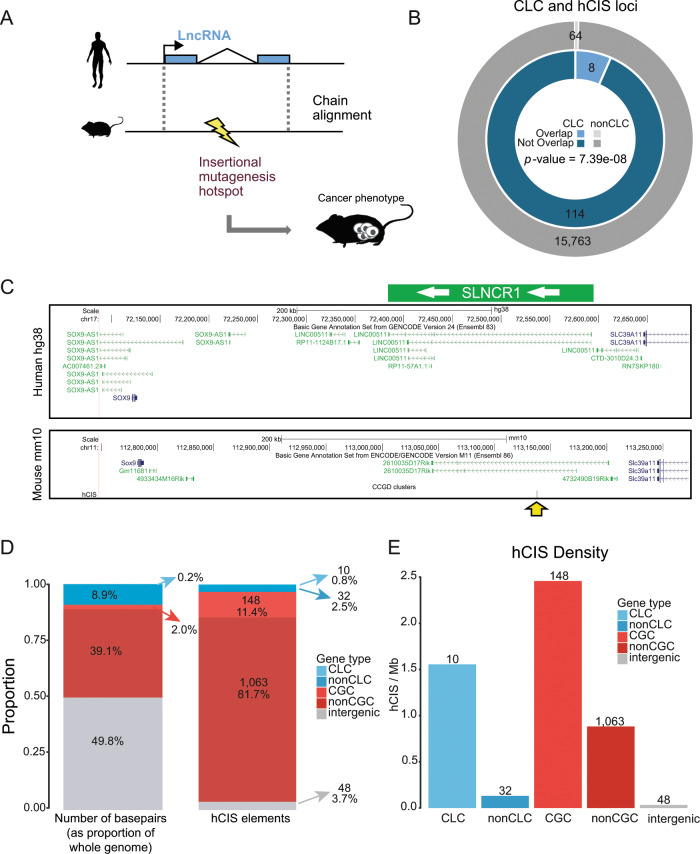
Long non-coding RNAs (lncRNAs) are a growing focus of cancer genomics studies, creating the need for a resource of lncRNAs with validated cancer roles. Furthermore, it remains debated whether mutated lncRNAs can drive tumorigenesis, and whether such functions could be conserved during evolution. Here, as part of the ICGC/TCGA Pan-Cancer Analysis of Whole Genomes (PCAWG) Consortium, we introduce the Cancer LncRNA Census (CLC), a compilation of 122 GENCODE lncRNAs with causal roles in cancer phenotypes. In contrast to existing databases, CLC requires strong functional or genetic evidence. CLC genes are enriched amongst driver genes predicted from somatic mutations, and display characteristic genomic features. Strikingly, CLC genes are enriched for driver mutations from unbiased, genome-wide transposon-mutagenesis screens in mice. We identified 10 tumour-causing mutations in orthologues of 8 lncRNAs, including LINC-PINT and NEAT1, but not MALAT1. Thus CLC represents a dataset of high-confidence cancer lncRNAs. Mutagenesis maps are a novel means for identifying deeply-conserved roles of lncRNAs in tumorigenesis.
Conflict of interest statement
The authors declare no competing interests.
Figures