

Pan-cancer analysis of whole genomes identifies driver rearrangements promoted by LINE-1 retrotransposition
- PMID: 32024998
- PMCID: PMC7058536
- DOI: 10.1038/s41588-019-0562-0
Pan-cancer analysis of whole genomes identifies driver rearrangements promoted by LINE-1 retrotransposition
Erratum in
-
Author Correction: Pan-cancer analysis of whole genomes identifies driver rearrangements promoted by LINE-1 retrotransposition.Nat Genet. 2023 Jun;55(6):1080. doi: 10.1038/s41588-023-01319-9. Nat Genet. 2023. PMID: 36944736 Free PMC article. No abstract available.
Abstract
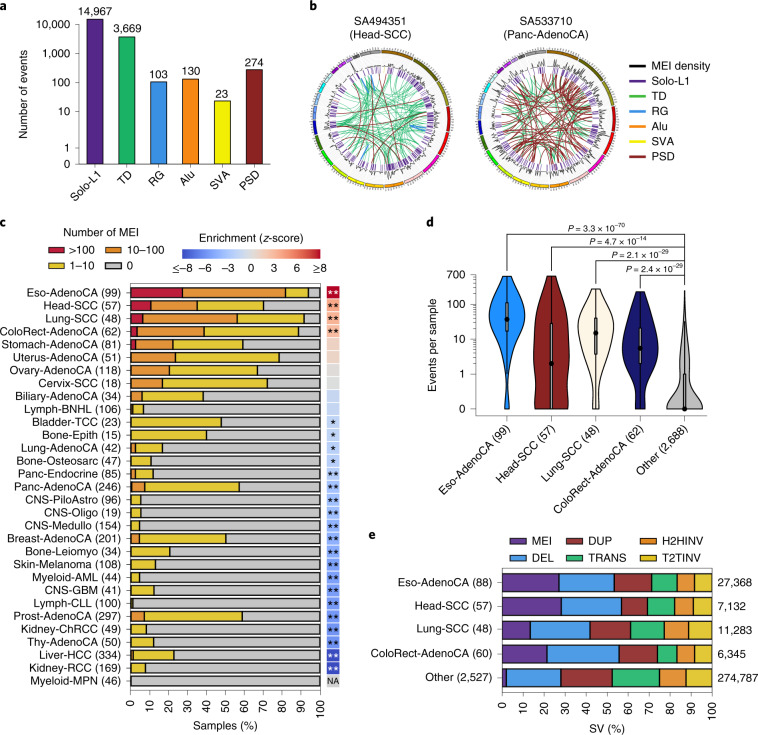
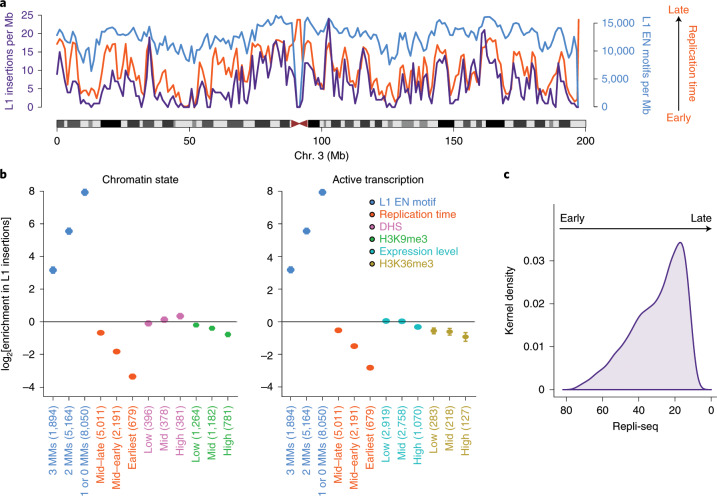
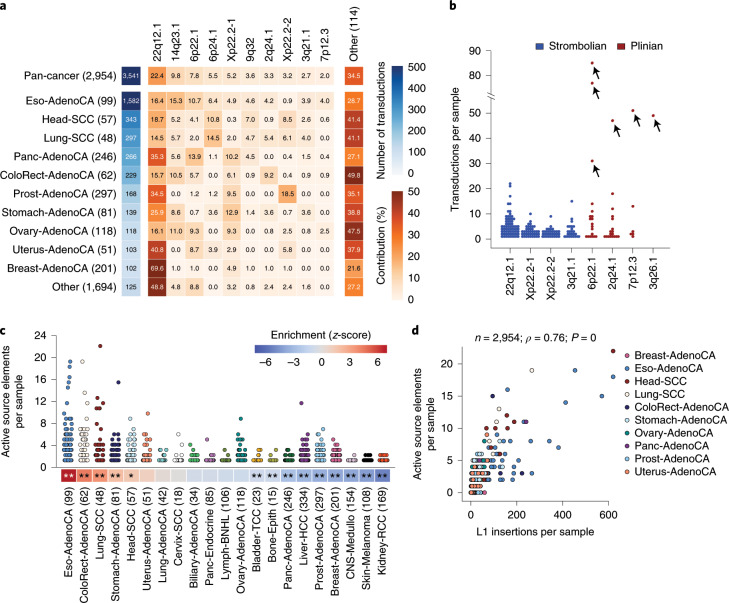
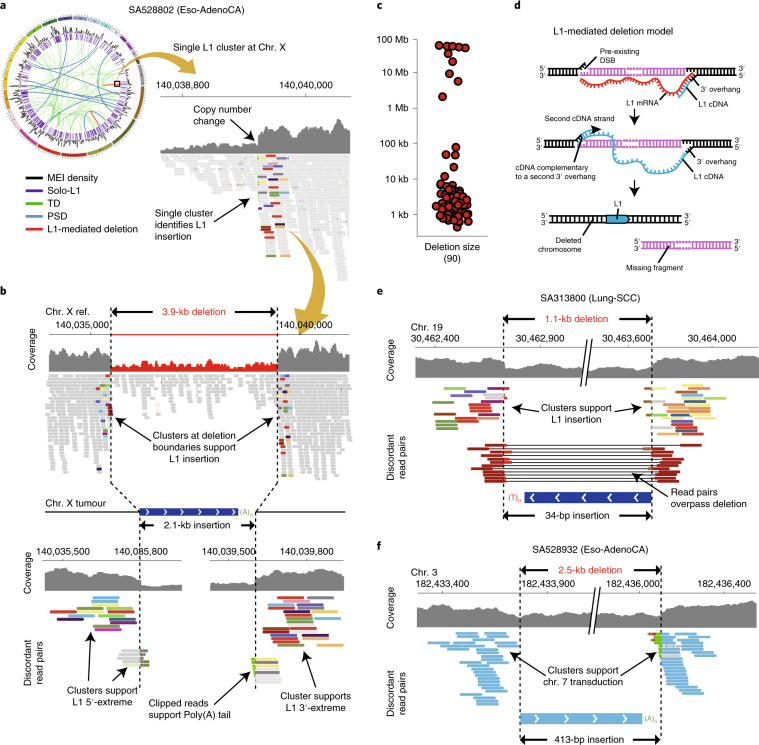
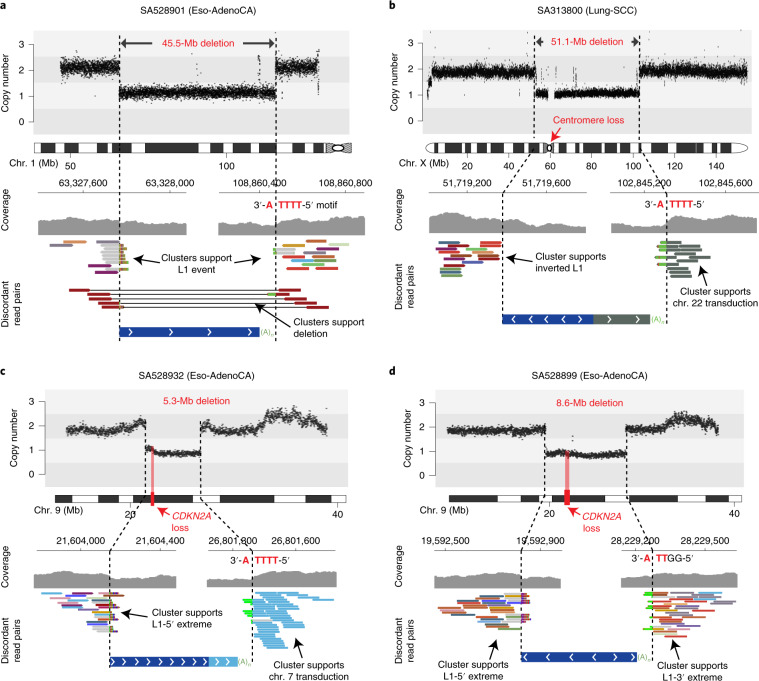
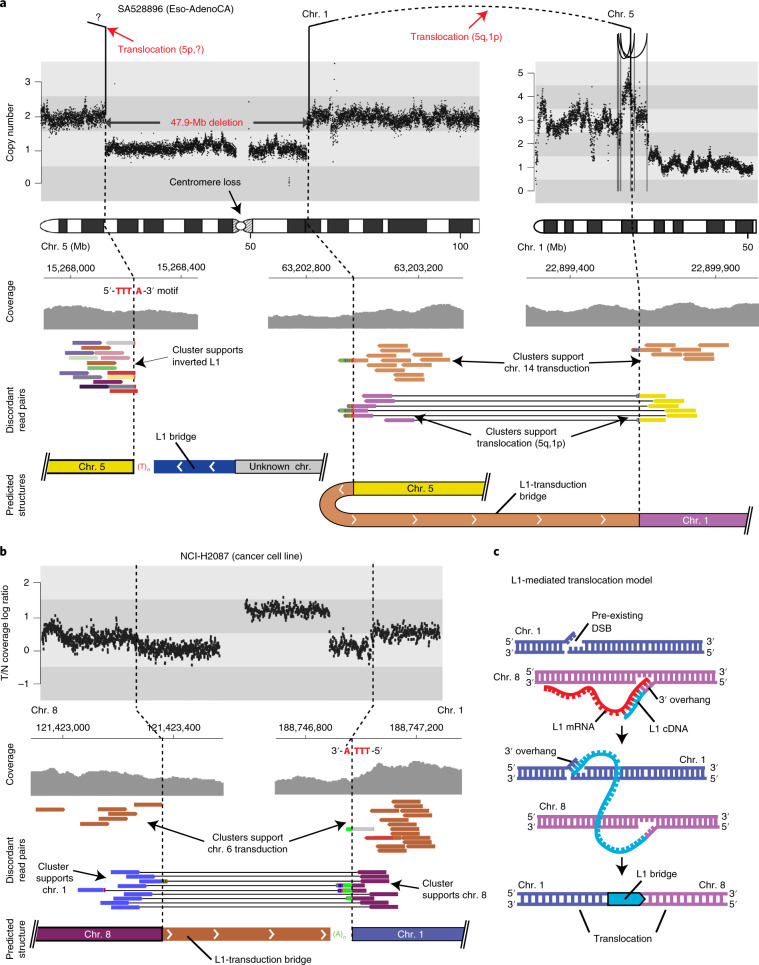
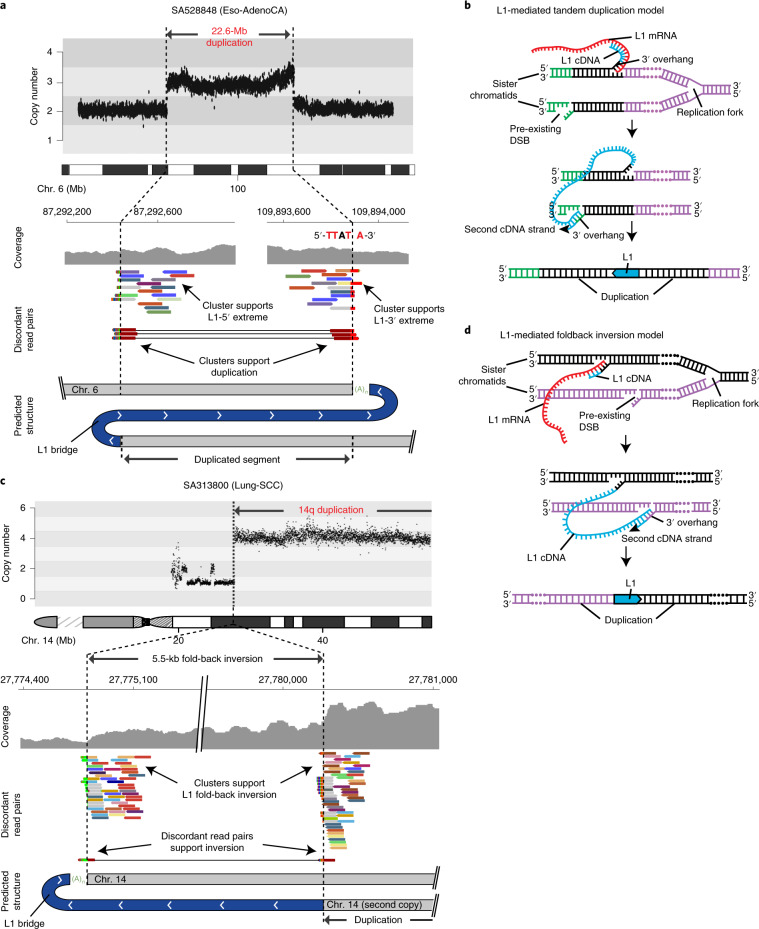
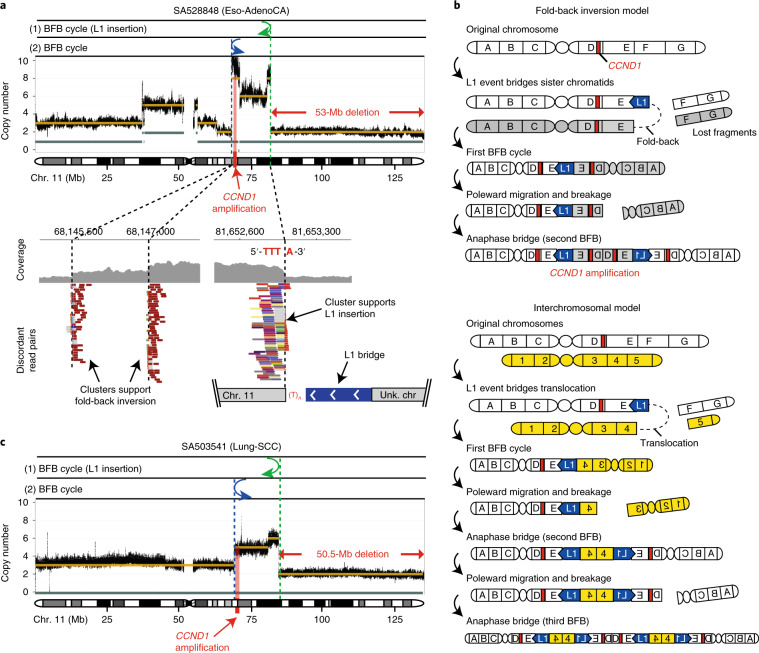
About half of all cancers have somatic integrations of retrotransposons. Here, to characterize their role in oncogenesis, we analyzed the patterns and mechanisms of somatic retrotransposition in 2,954 cancer genomes from 38 histological cancer subtypes within the framework of the Pan-Cancer Analysis of Whole Genomes (PCAWG) project. We identified 19,166 somatically acquired retrotransposition events, which affected 35% of samples and spanned a range of event types. Long interspersed nuclear element (LINE-1; L1 hereafter) insertions emerged as the first most frequent type of somatic structural variation in esophageal adenocarcinoma, and the second most frequent in head-and-neck and colorectal cancers. Aberrant L1 integrations can delete megabase-scale regions of a chromosome, which sometimes leads to the removal of tumor-suppressor genes, and can induce complex translocations and large-scale duplications. Somatic retrotranspositions can also initiate breakage-fusion-bridge cycles, leading to high-level amplification of oncogenes. These observations illuminate a relevant role of L1 retrotransposition in remodeling the cancer genome, with potential implications for the development of human tumors.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
- R01 CA188228/CA/NCI NIH HHS/United States
- R01 CA172652/CA/NCI NIH HHS/United States
- 23924/CRUK_/Cancer Research UK/United Kingdom
- 22720/CRUK_/Cancer Research UK/United Kingdom
- T32 CA009110/CA/NCI NIH HHS/United States
- T32 HG002295/HG/NHGRI NIH HHS/United States
- R01 GM124531/GM/NIGMS NIH HHS/United States
- 088340/WT_/Wellcome Trust/United Kingdom
- 093867/WT_/Wellcome Trust/United Kingdom
- R01 CA217991/CA/NCI NIH HHS/United States
- K01 AG051791/AG/NIA NIH HHS/United States
- 14545/CRUK_/Cancer Research UK/United Kingdom
- U24 CA210978/CA/NCI NIH HHS/United States
- FC001202/WT_/Wellcome Trust/United Kingdom
- R35 GM127029/GM/NIGMS NIH HHS/United States
- P30 ES010126/ES/NIEHS NIH HHS/United States
- P30 CA016672/CA/NCI NIH HHS/United States
- R01 GM099875/GM/NIGMS NIH HHS/United States
- 21777/CRUK_/Cancer Research UK/United Kingdom
- R01 CA163705/CA/NCI NIH HHS/United States
- R01 CA095175/CA/NCI NIH HHS/United States
- U41 HG007497/HG/NHGRI NIH HHS/United States
- 757700/ERC_/European Research Council/International
- 155365/SNSF_/Swiss National Science Foundation/Switzerland
- A21777/CRUK_/Cancer Research UK/United Kingdom
- P50 GM107632/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
