

Disruption of chromatin folding domains by somatic genomic rearrangements in human cancer
- PMID: 32024999
- PMCID: PMC7058537
- DOI: 10.1038/s41588-019-0564-y
Disruption of chromatin folding domains by somatic genomic rearrangements in human cancer
Erratum in
-
Author Correction: Disruption of chromatin folding domains by somatic genomic rearrangements in human cancer.Nat Genet. 2023 Jun;55(6):1079. doi: 10.1038/s41588-023-01318-w. Nat Genet. 2023. PMID: 36944735 Free PMC article. No abstract available.
Abstract
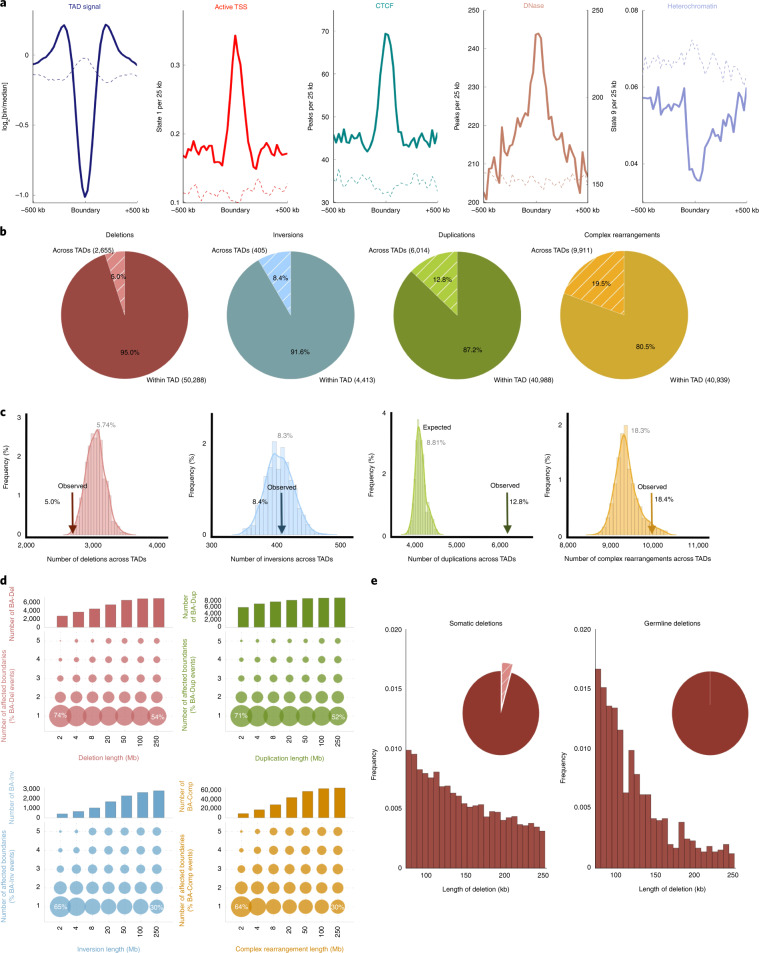
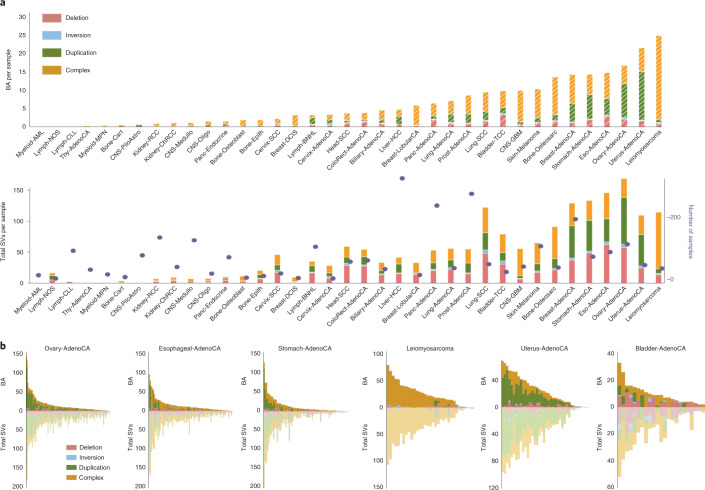
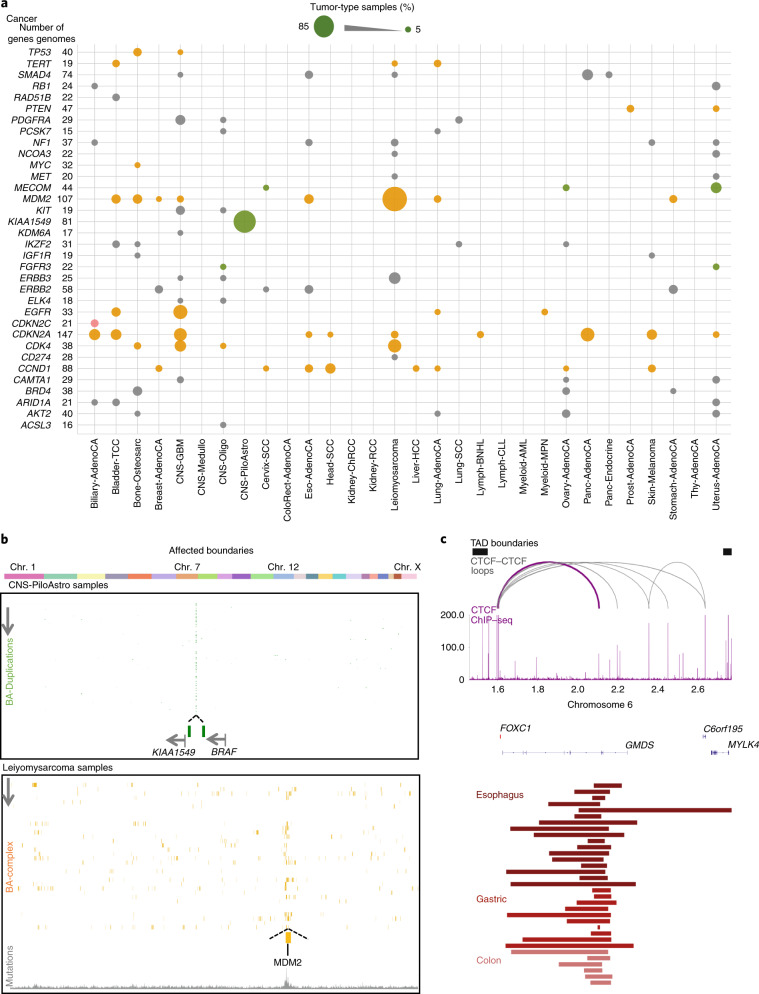
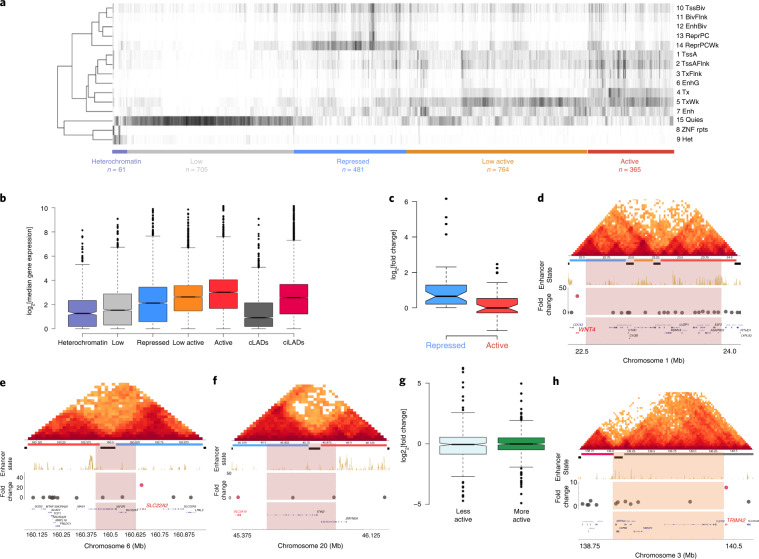
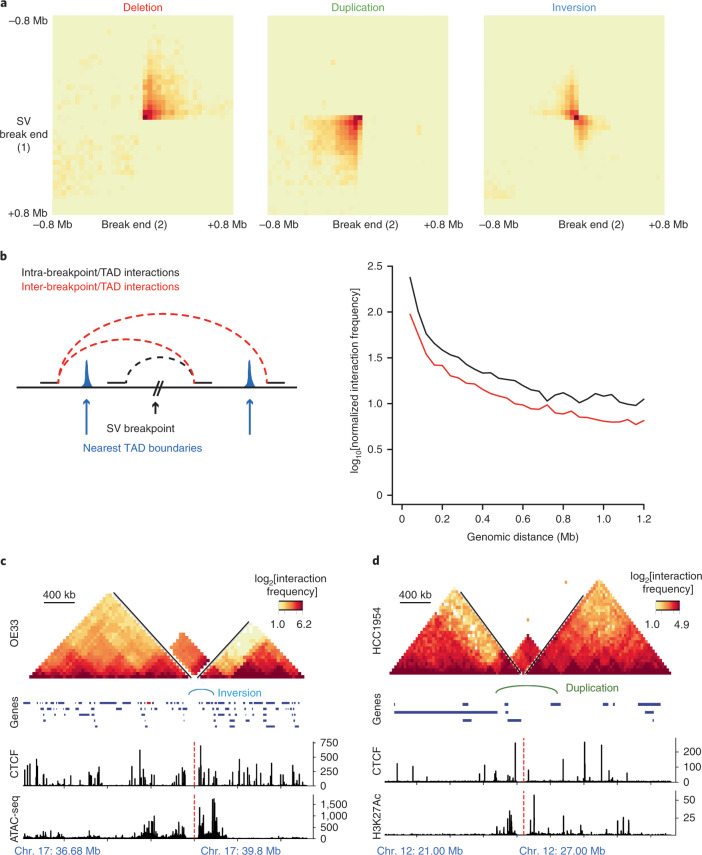
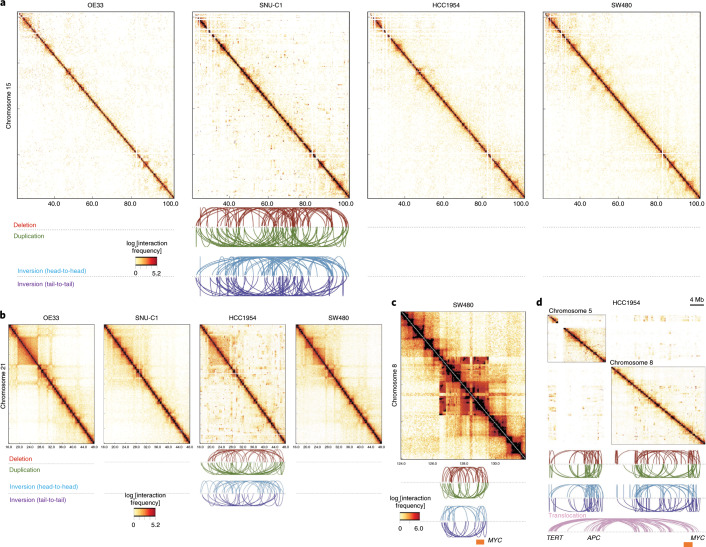
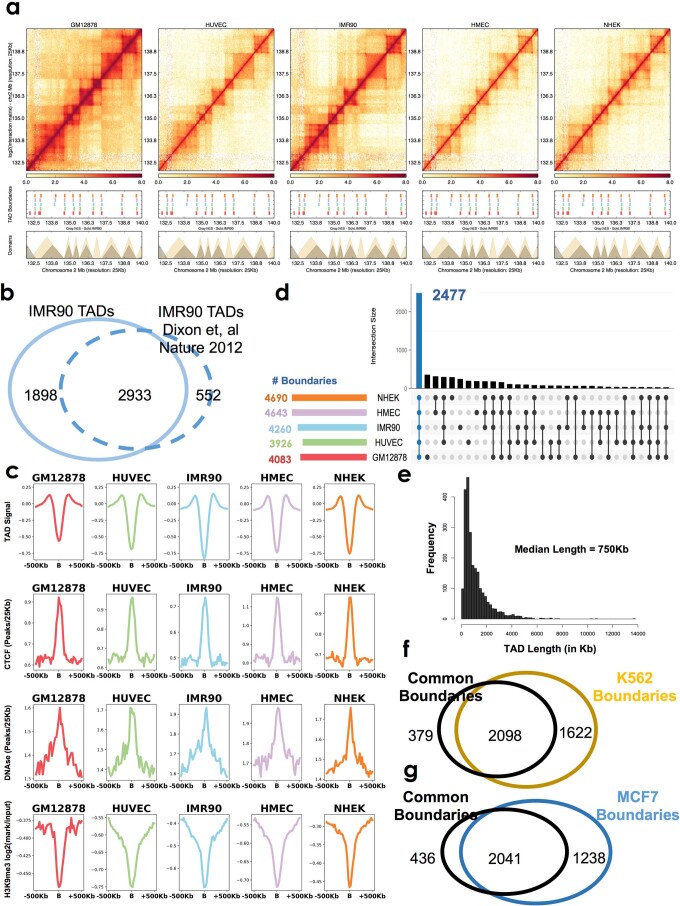
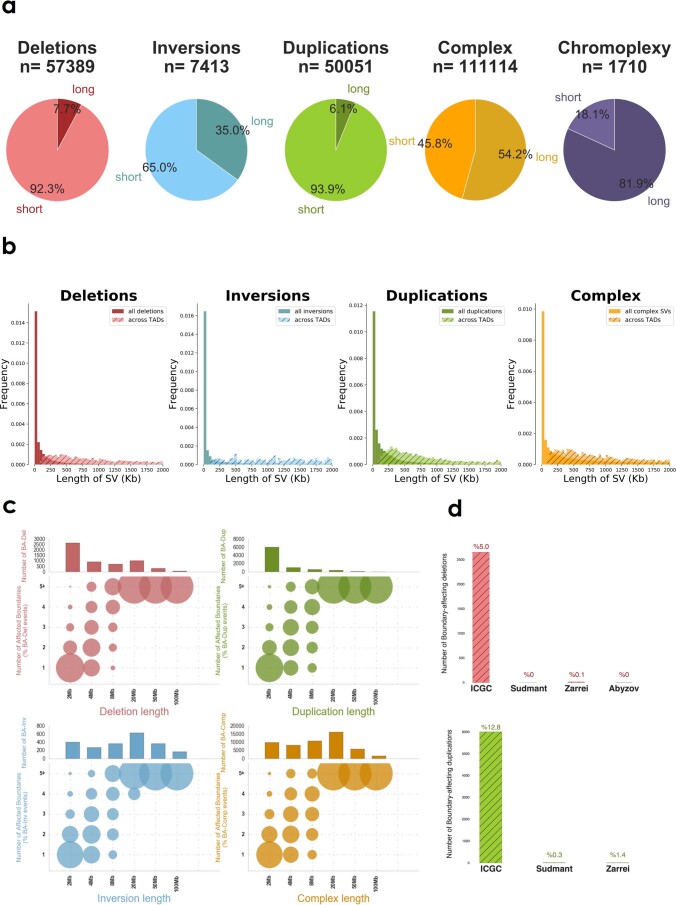
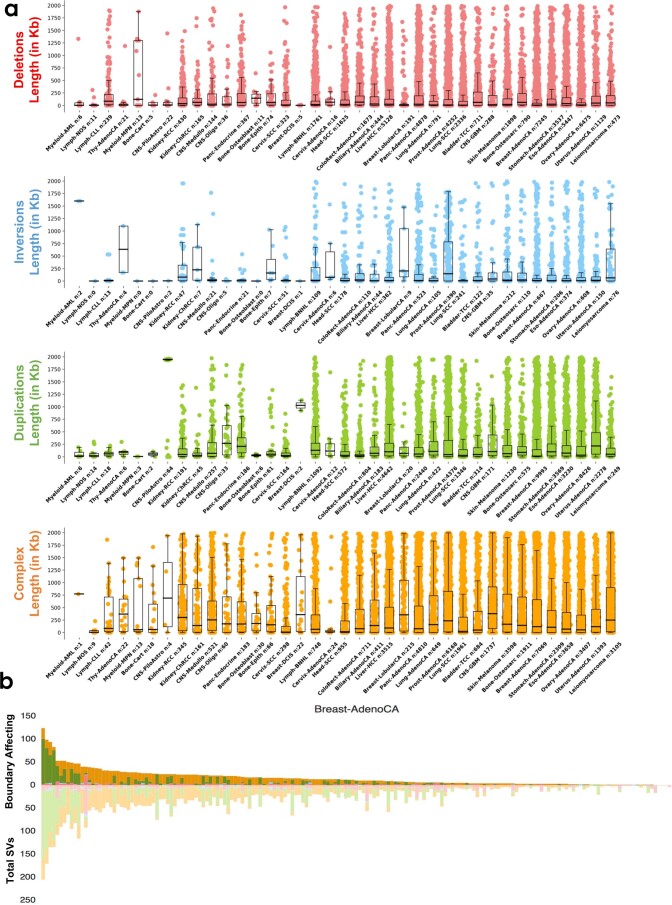
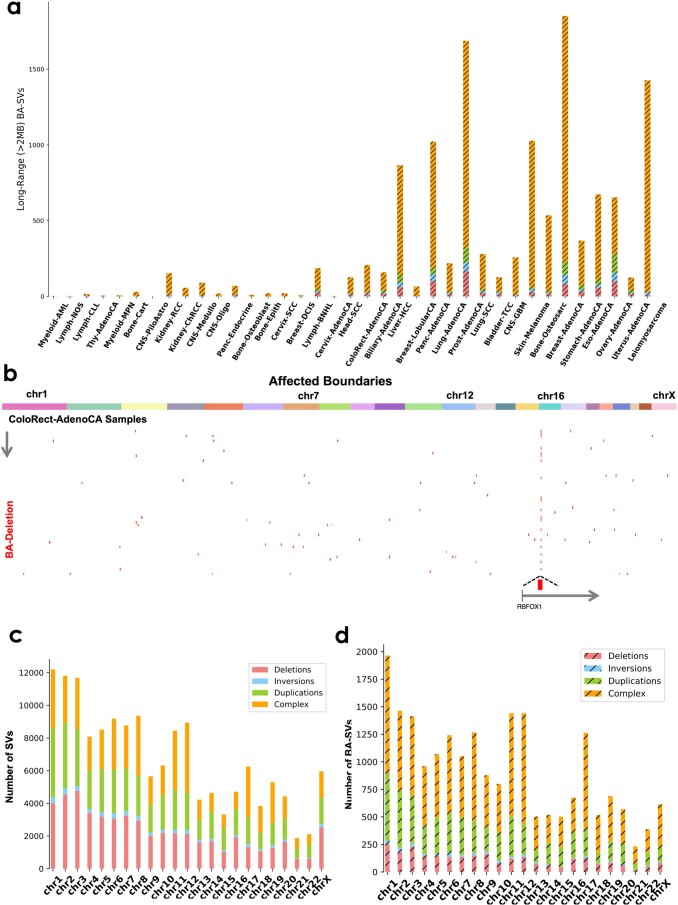
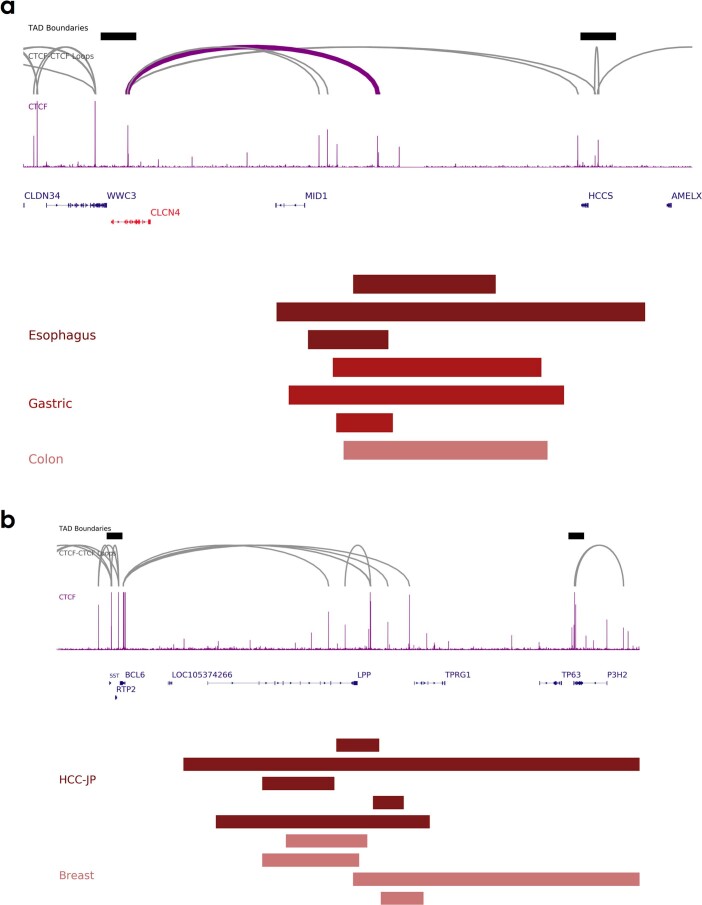
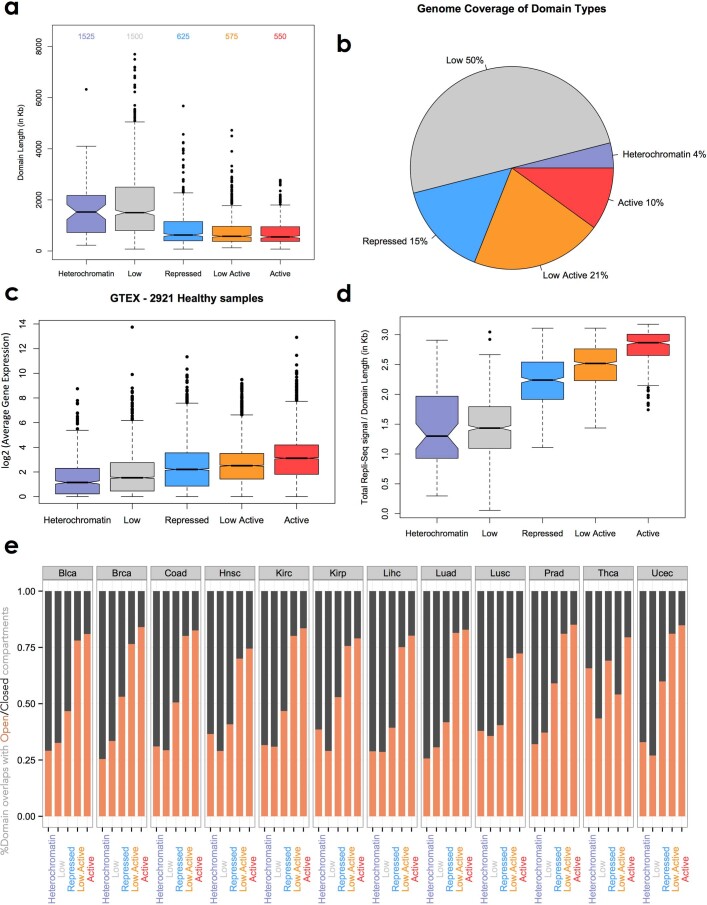
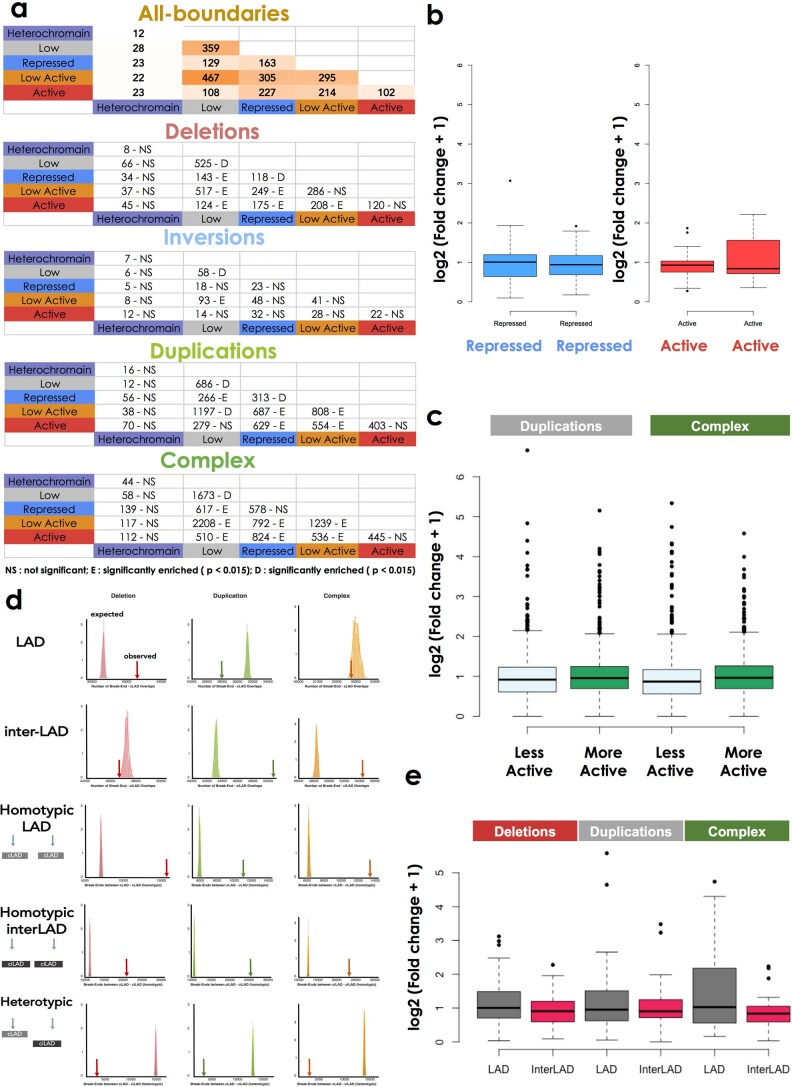
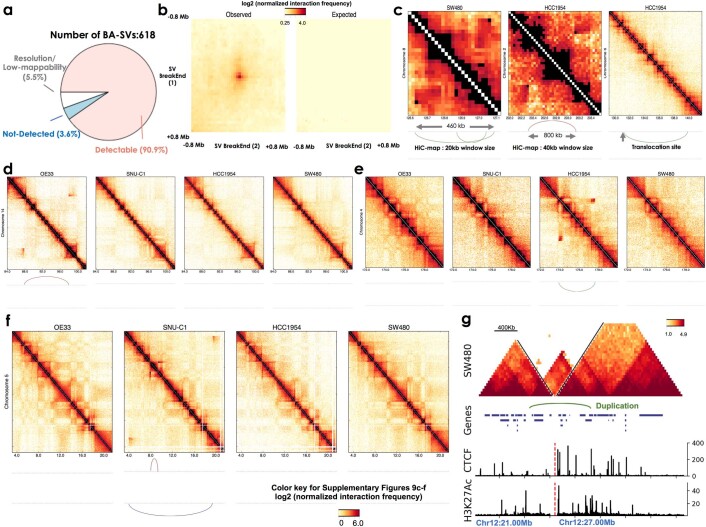
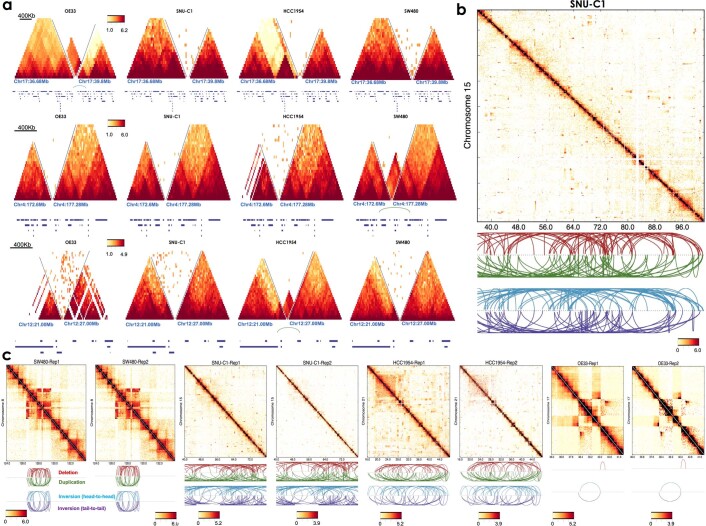
Chromatin is folded into successive layers to organize linear DNA. Genes within the same topologically associating domains (TADs) demonstrate similar expression and histone-modification profiles, and boundaries separating different domains have important roles in reinforcing the stability of these features. Indeed, domain disruptions in human cancers can lead to misregulation of gene expression. However, the frequency of domain disruptions in human cancers remains unclear. Here, as part of the Pan-Cancer Analysis of Whole Genomes (PCAWG) Consortium of the International Cancer Genome Consortium (ICGC) and The Cancer Genome Atlas (TCGA), which aggregated whole-genome sequencing data from 2,658 cancers across 38 tumor types, we analyzed 288,457 somatic structural variations (SVs) to understand the distributions and effects of SVs across TADs. Notably, SVs can lead to the fusion of discrete TADs, and complex rearrangements markedly change chromatin folding maps in the cancer genomes. Notably, only 14% of the boundary deletions resulted in a change in expression in nearby genes of more than twofold.
Conflict of interest statement
R.B. owns equity in Ampressa Therapeutics, is the chair of the scientific advisory board of and consultant for OrigiMed, has received research funding from Bayer and Ono Pharma, and receives patent royalties from LabCorp. All other authors have no competing interests.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
