

Genomic basis for RNA alterations in cancer
- PMID: 32025019
- PMCID: PMC7054216
- DOI: 10.1038/s41586-020-1970-0
Genomic basis for RNA alterations in cancer
Erratum in
-
Author Correction: Genomic basis for RNA alterations in cancer.Nature. 2023 Feb;614(7948):E37. doi: 10.1038/s41586-022-05596-y. Nature. 2023. PMID: 36697831 Free PMC article. No abstract available.
Abstract
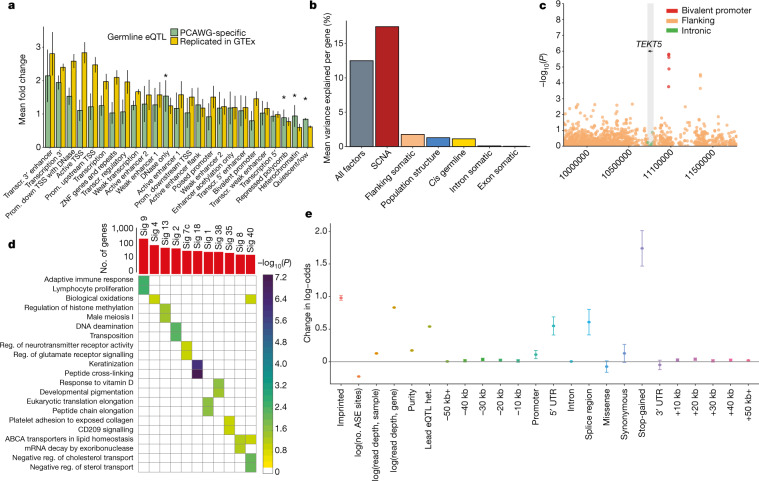
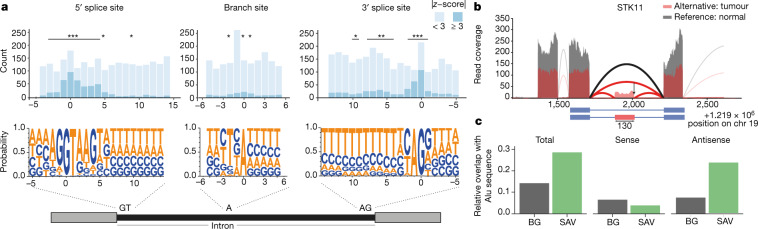
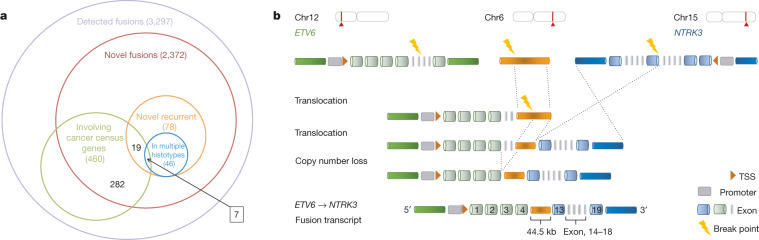
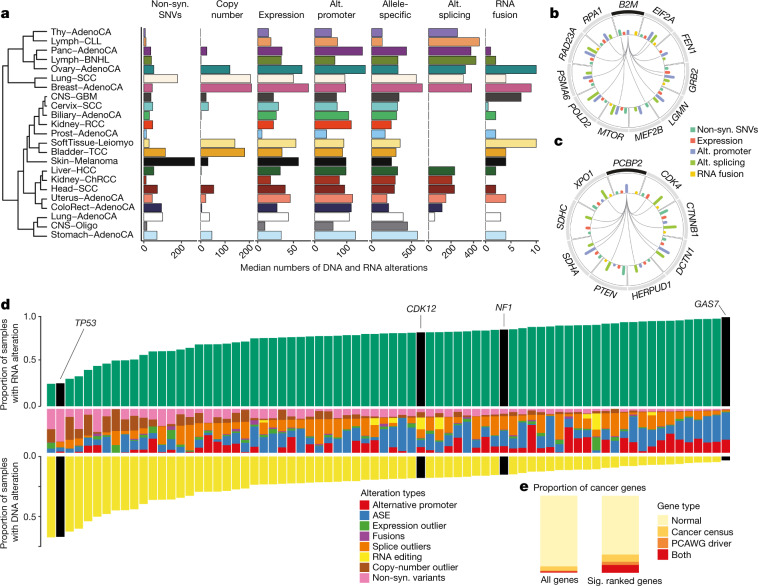
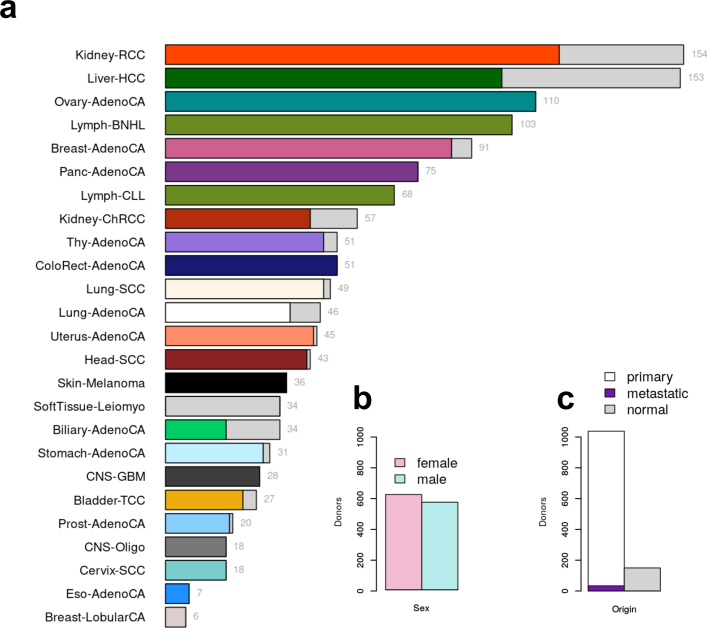
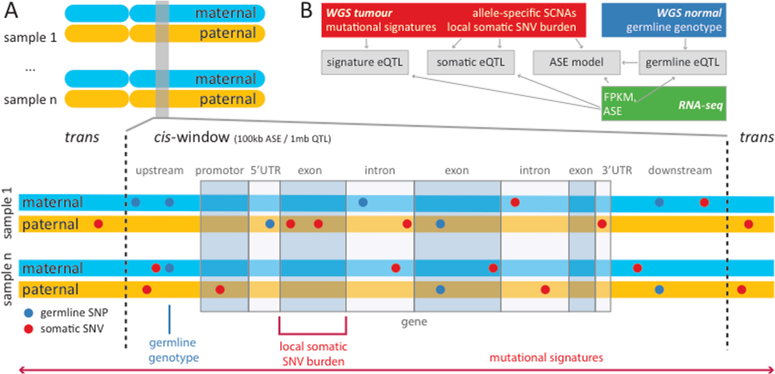
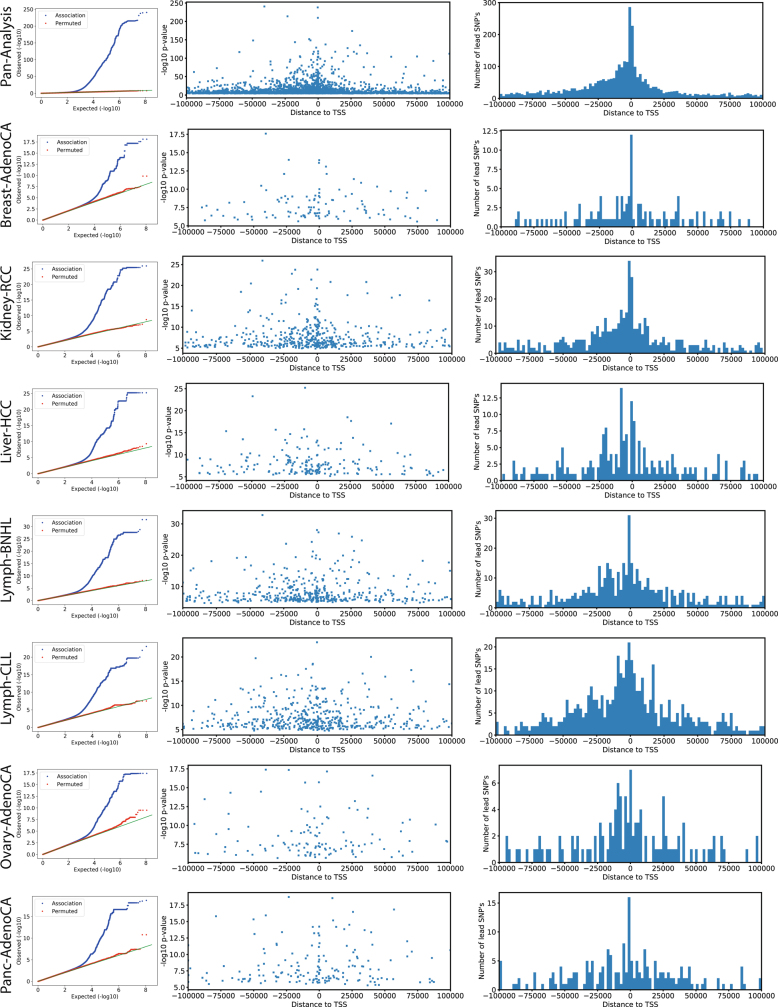
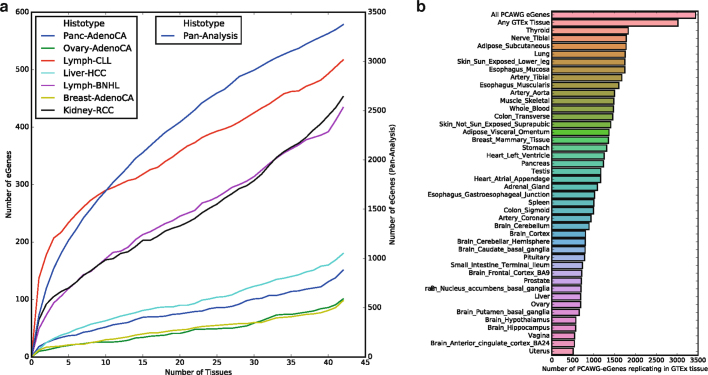
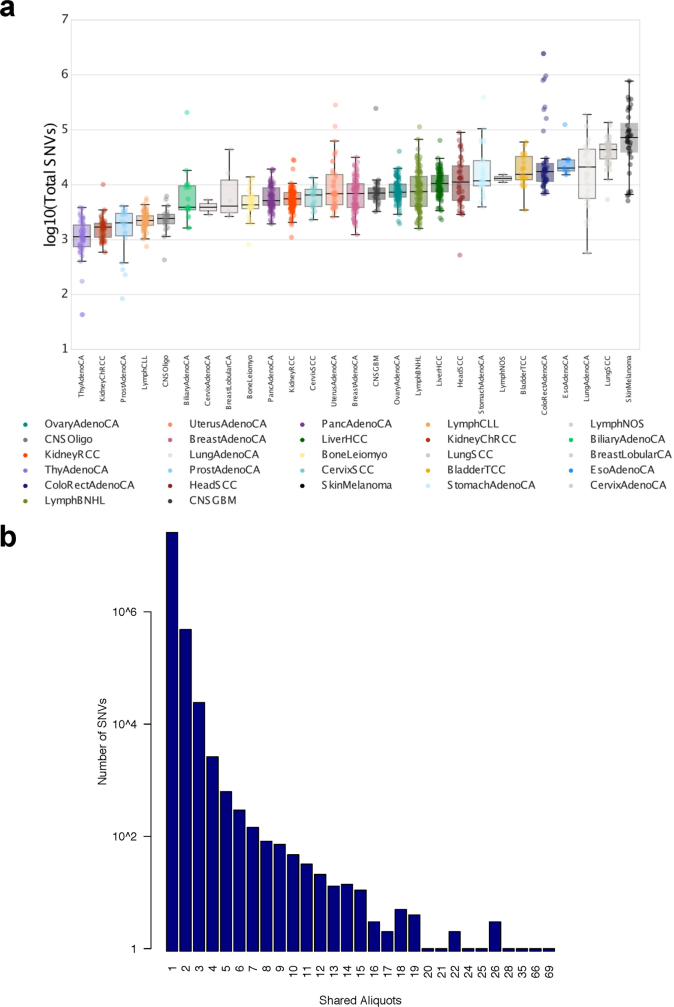
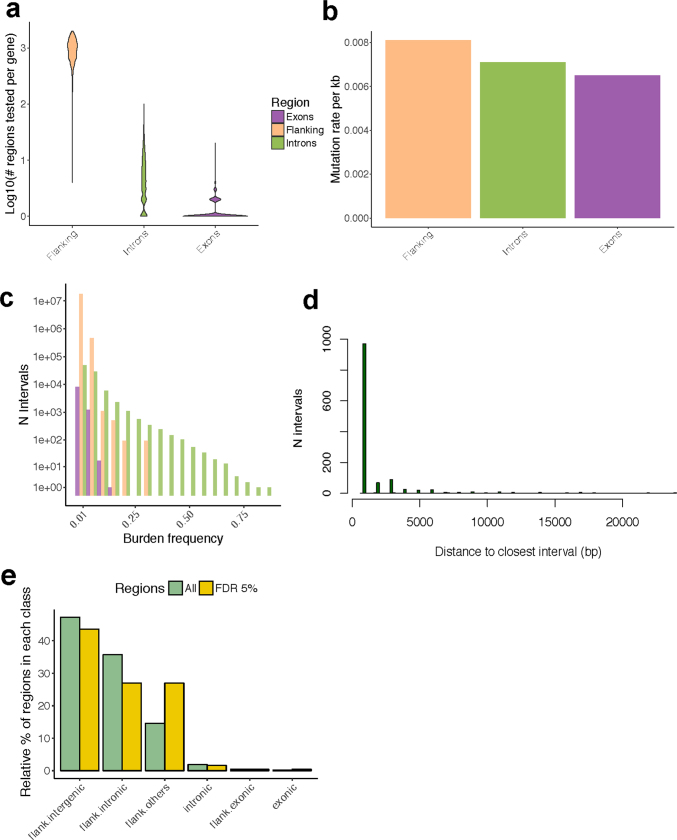
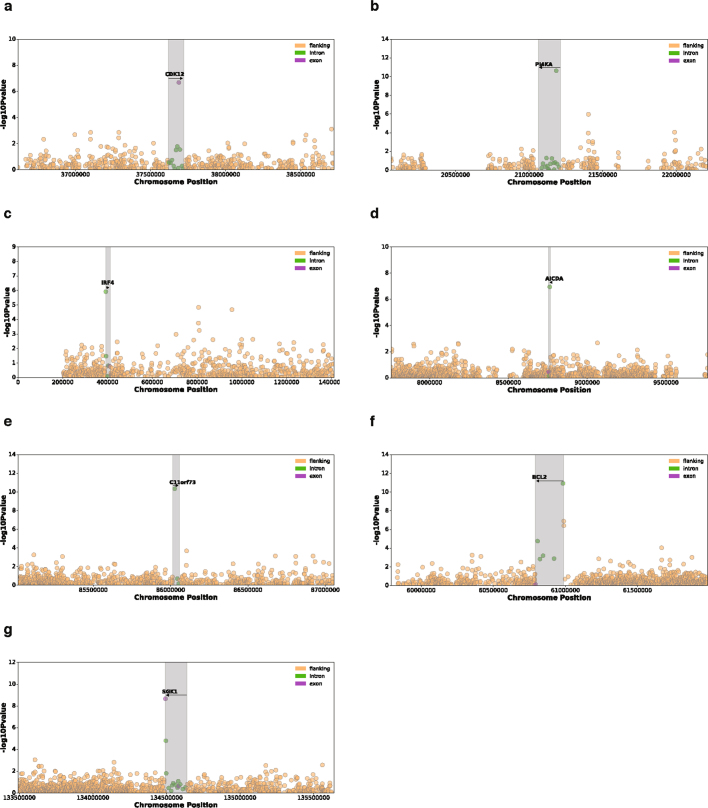
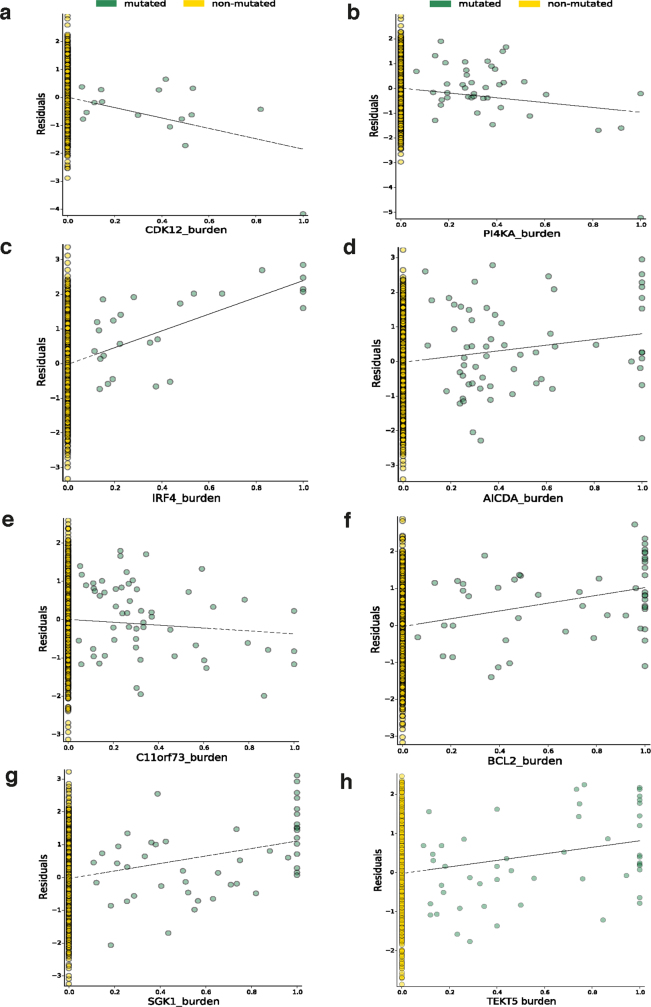
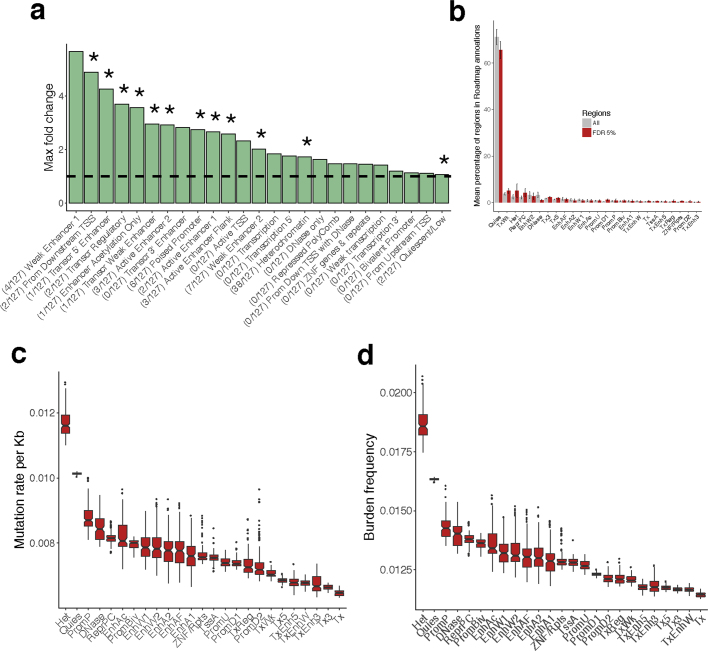
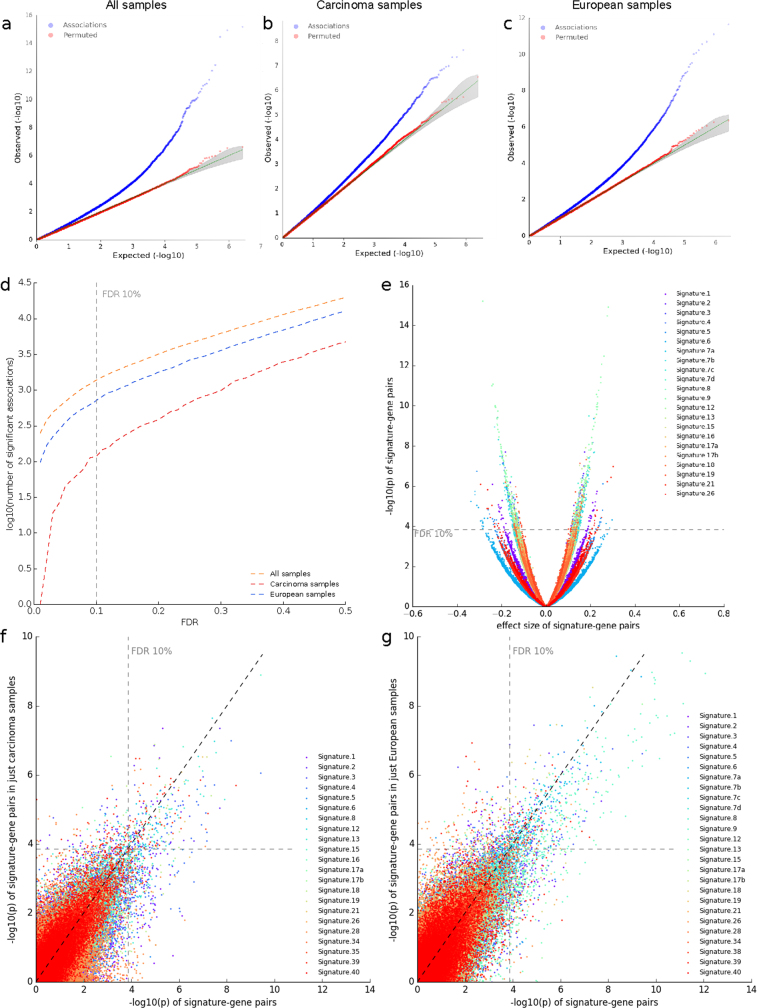
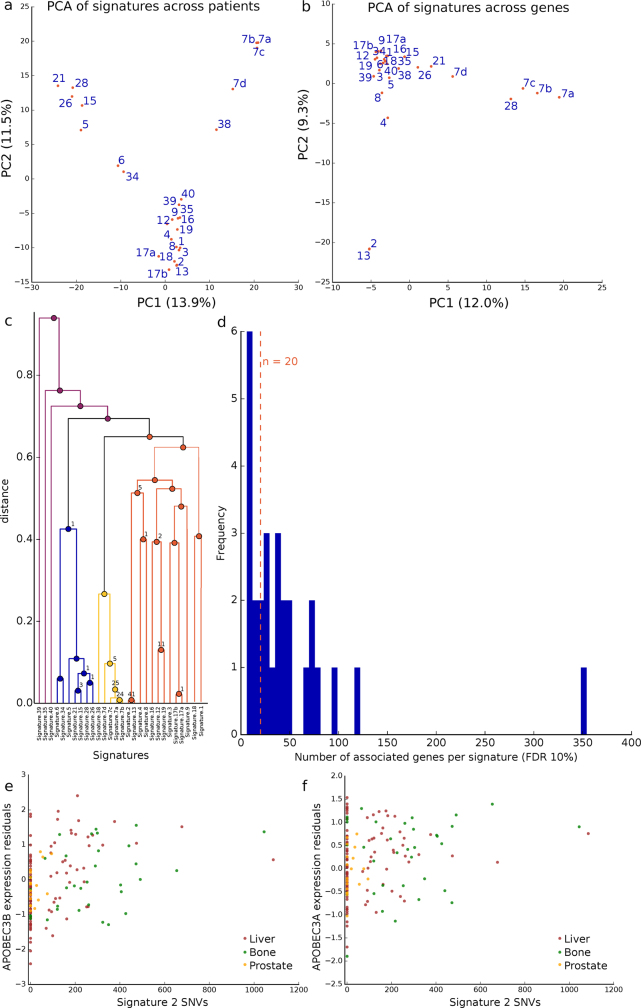
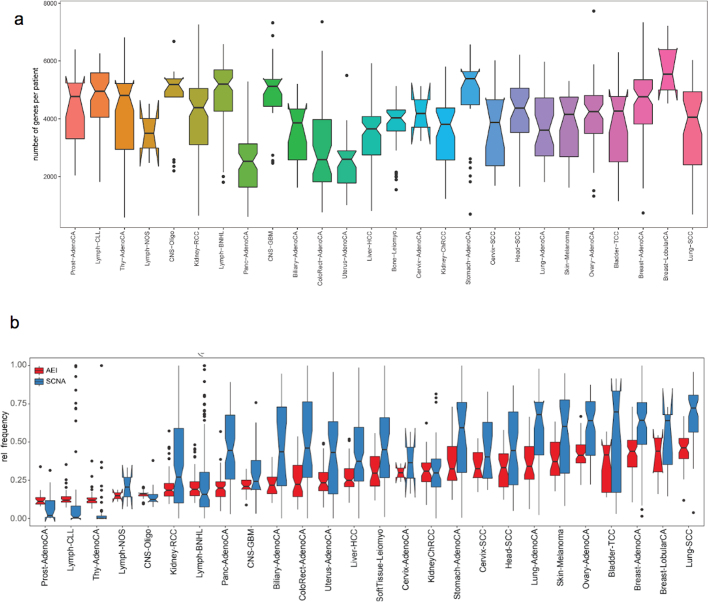
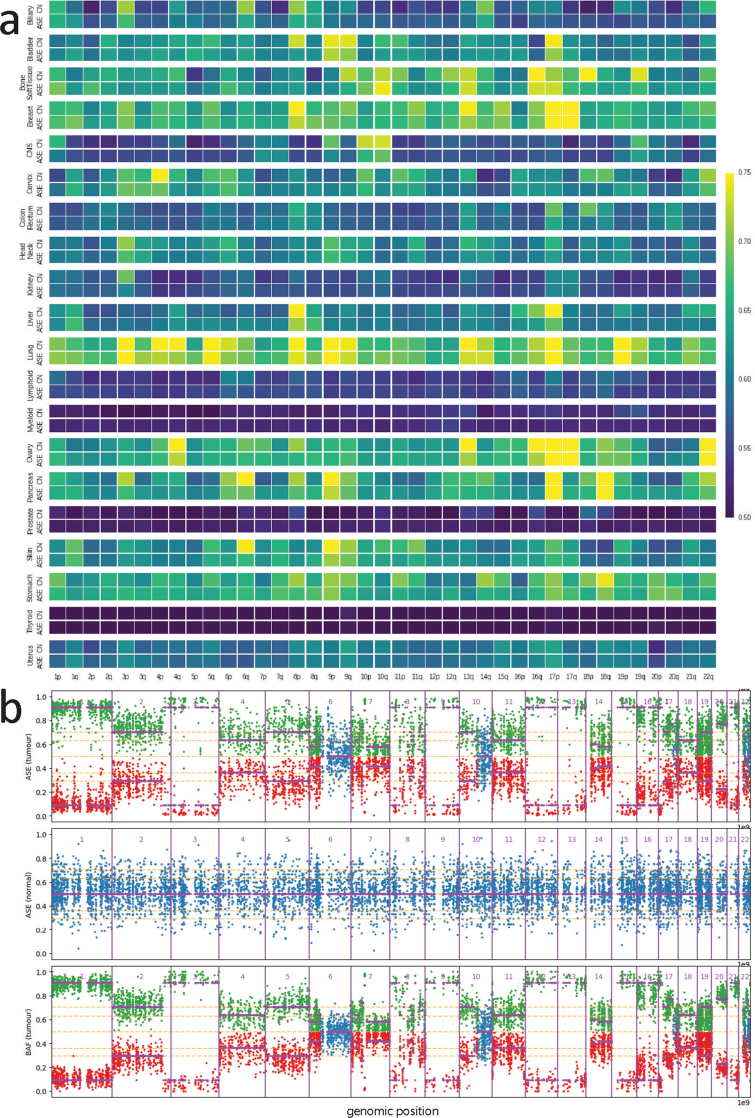
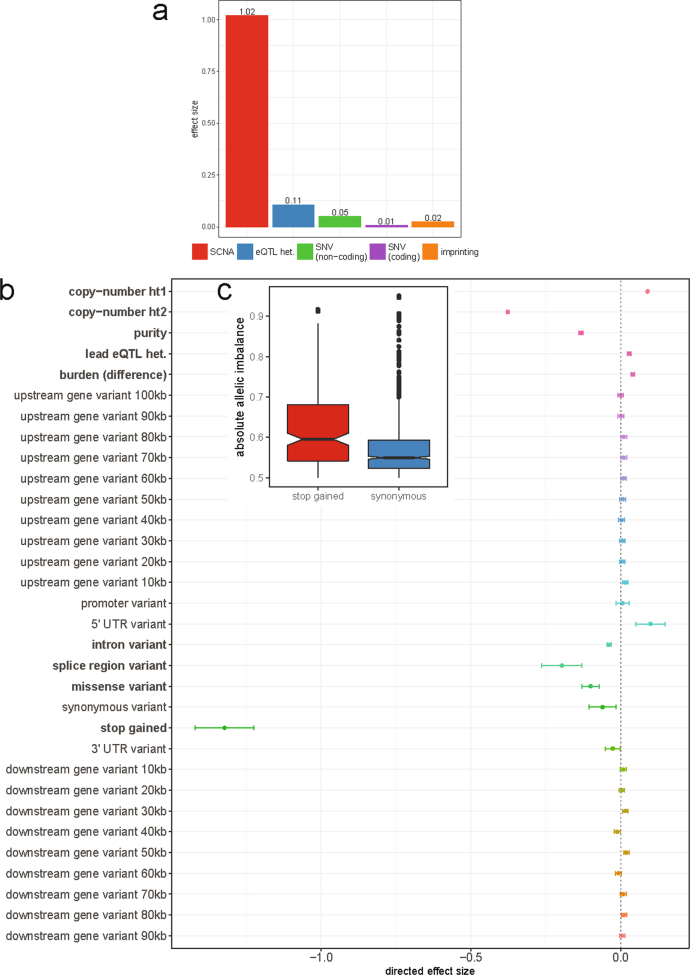
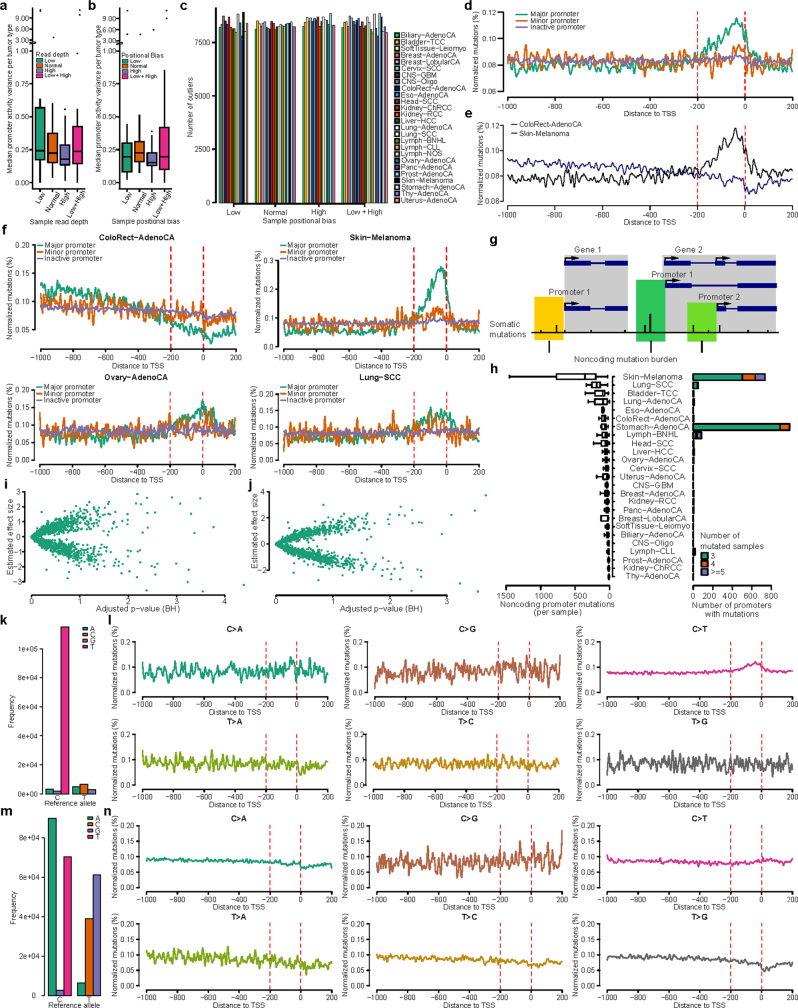
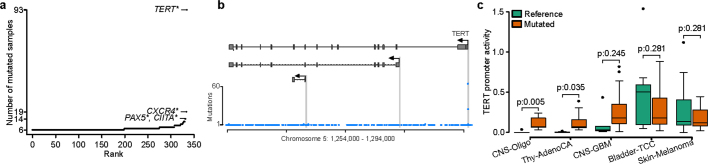
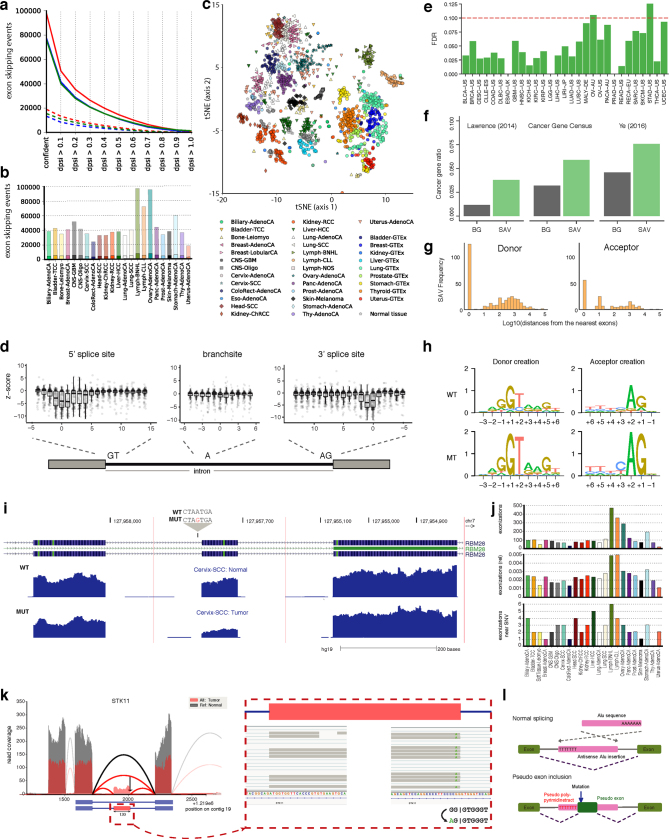
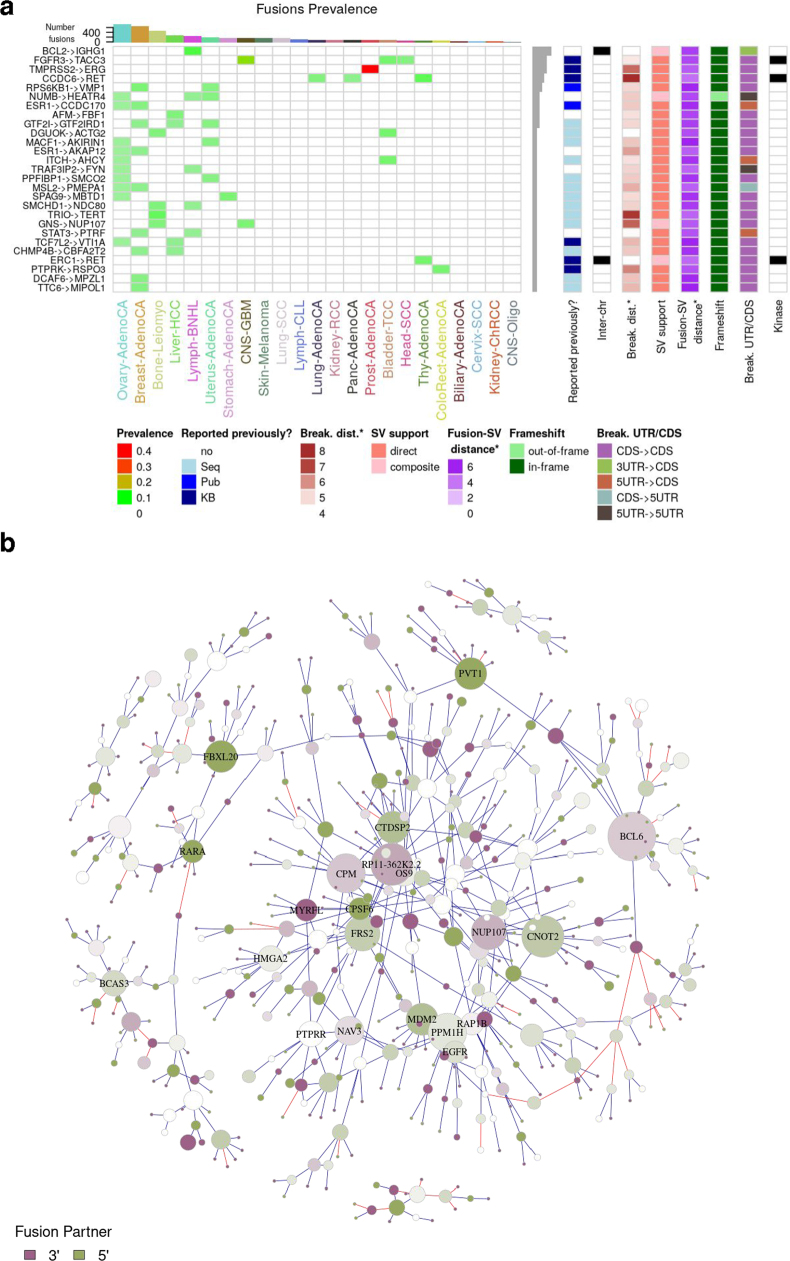
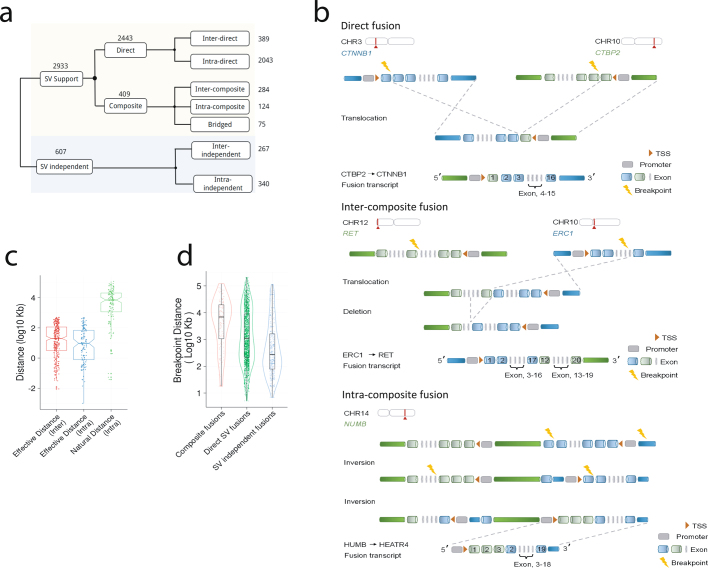
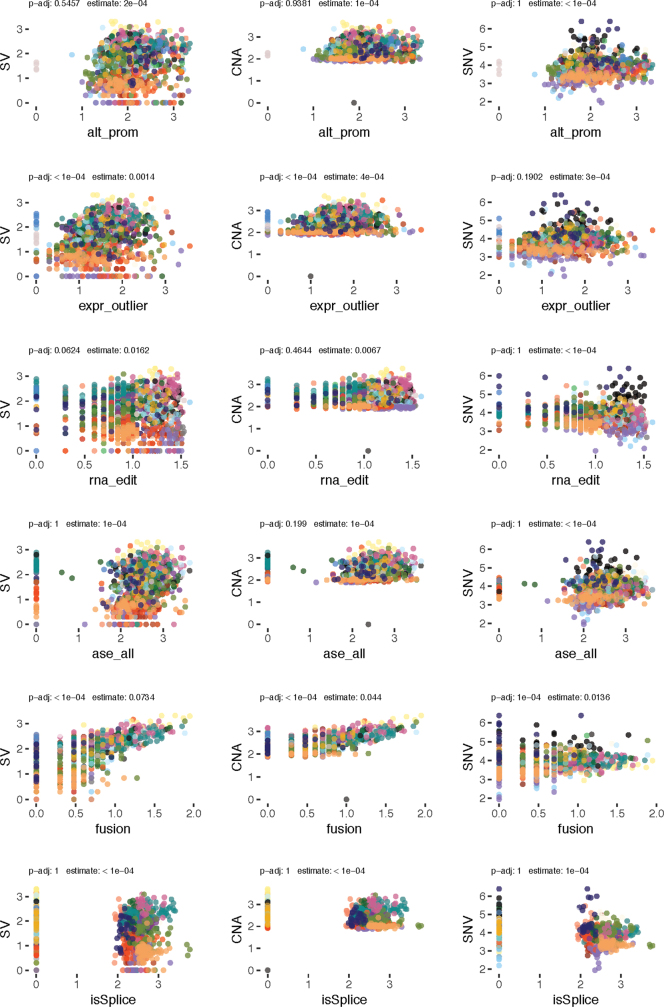
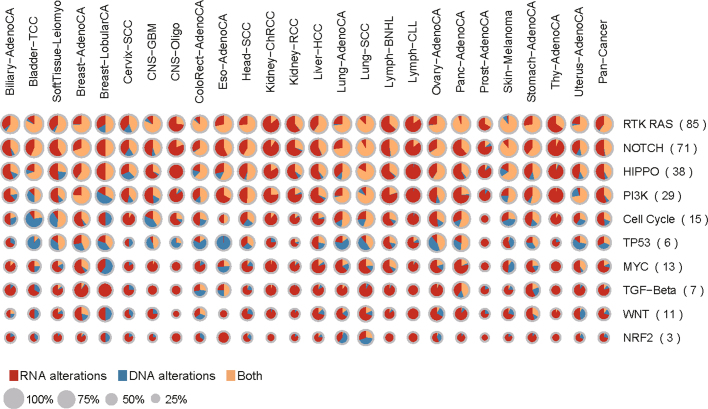
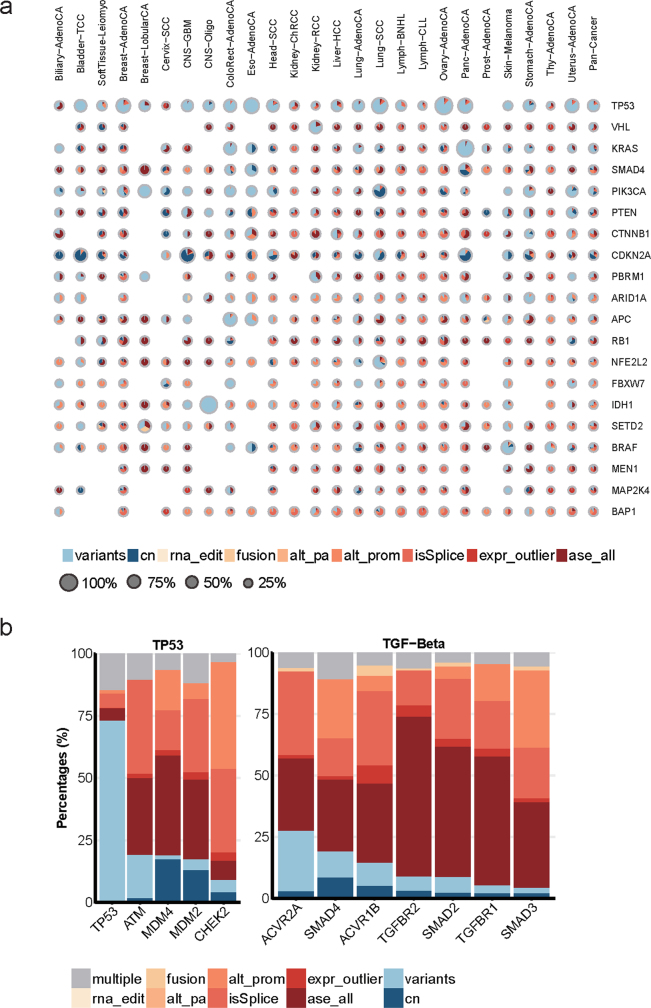
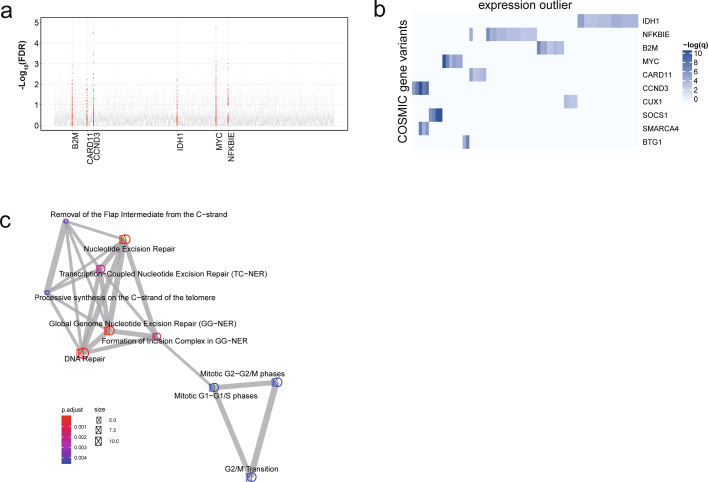
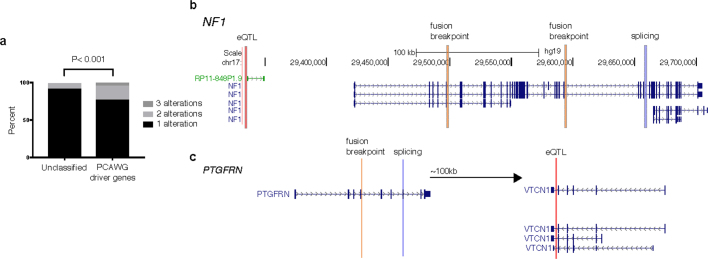
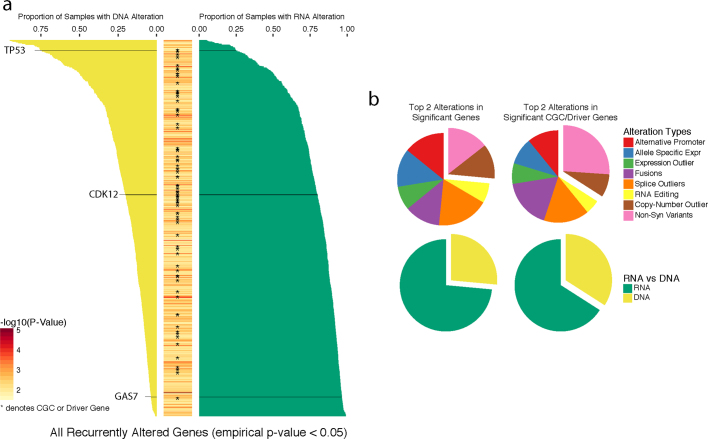
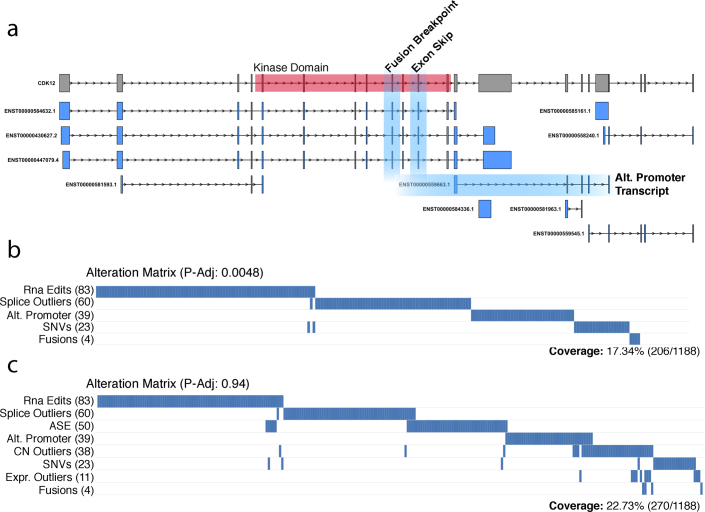
Transcript alterations often result from somatic changes in cancer genomes1. Various forms of RNA alterations have been described in cancer, including overexpression2, altered splicing3 and gene fusions4; however, it is difficult to attribute these to underlying genomic changes owing to heterogeneity among patients and tumour types, and the relatively small cohorts of patients for whom samples have been analysed by both transcriptome and whole-genome sequencing. Here we present, to our knowledge, the most comprehensive catalogue of cancer-associated gene alterations to date, obtained by characterizing tumour transcriptomes from 1,188 donors of the Pan-Cancer Analysis of Whole Genomes (PCAWG) Consortium of the International Cancer Genome Consortium (ICGC) and The Cancer Genome Atlas (TCGA)5. Using matched whole-genome sequencing data, we associated several categories of RNA alterations with germline and somatic DNA alterations, and identified probable genetic mechanisms. Somatic copy-number alterations were the major drivers of variations in total gene and allele-specific expression. We identified 649 associations of somatic single-nucleotide variants with gene expression in cis, of which 68.4% involved associations with flanking non-coding regions of the gene. We found 1,900 splicing alterations associated with somatic mutations, including the formation of exons within introns in proximity to Alu elements. In addition, 82% of gene fusions were associated with structural variants, including 75 of a new class, termed 'bridged' fusions, in which a third genomic location bridges two genes. We observed transcriptomic alteration signatures that differ between cancer types and have associations with variations in DNA mutational signatures. This compendium of RNA alterations in the genomic context provides a rich resource for identifying genes and mechanisms that are functionally implicated in cancer.
Conflict of interest statement
M.M. is a scientific advisory board chair of, and consultant for, OrigiMed, receives research funding from Bayer and Ono Pharma, and has patent royalties from LabCorp. G.R. is on the scientific advisory board of Computomics GmbH and receives research funding from Roche Diagnostics and Google. R.S. received honorariums for speaking at meeting organized by Roche and AstraZeneca. All the other authors have no competing interests.
Figures
Comment in
-
Global genomics project unravels cancer's complexity at unprecedented scale.Nature. 2020 Feb;578(7793):39-40. doi: 10.1038/d41586-020-00213-2. Nature. 2020. PMID: 32025004 No abstract available.
References
-
- The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Network. Pan-cancer analysis of whole genomes. Nature10.1038/s41586-020-1969-6 (2020).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
