Toward a gold standard for benchmarking gene set enrichment analysis
- PMID: 32026945
- PMCID: PMC7820859
- DOI: 10.1093/bib/bbz158
Toward a gold standard for benchmarking gene set enrichment analysis
Abstract
Motivation: Although gene set enrichment analysis has become an integral part of high-throughput gene expression data analysis, the assessment of enrichment methods remains rudimentary and ad hoc. In the absence of suitable gold standards, evaluations are commonly restricted to selected datasets and biological reasoning on the relevance of resulting enriched gene sets.
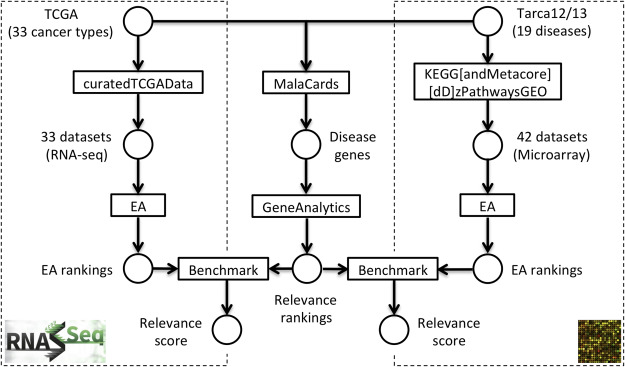
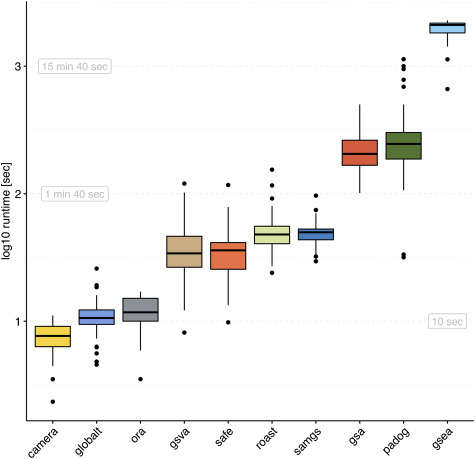
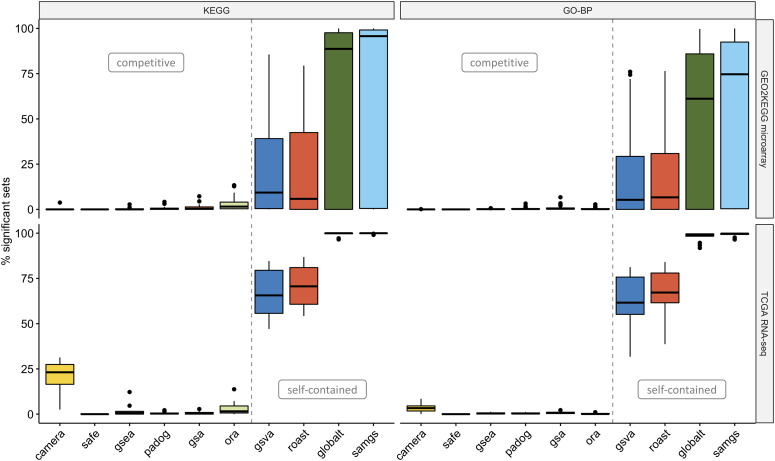
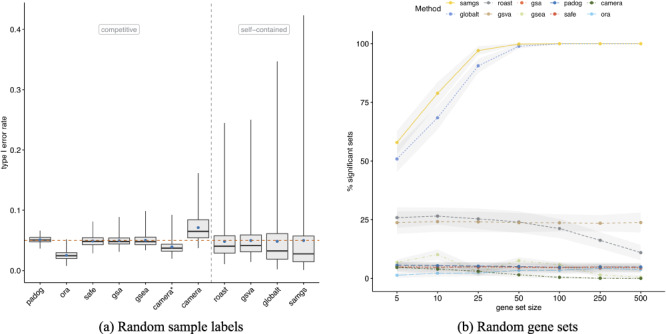
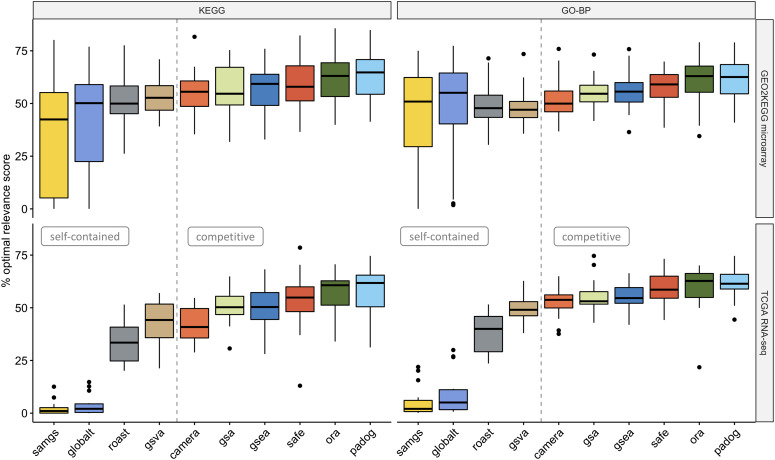
Results: We develop an extensible framework for reproducible benchmarking of enrichment methods based on defined criteria for applicability, gene set prioritization and detection of relevant processes. This framework incorporates a curated compendium of 75 expression datasets investigating 42 human diseases. The compendium features microarray and RNA-seq measurements, and each dataset is associated with a precompiled GO/KEGG relevance ranking for the corresponding disease under investigation. We perform a comprehensive assessment of 10 major enrichment methods, identifying significant differences in runtime and applicability to RNA-seq data, fraction of enriched gene sets depending on the null hypothesis tested and recovery of the predefined relevance rankings. We make practical recommendations on how methods originally developed for microarray data can efficiently be applied to RNA-seq data, how to interpret results depending on the type of gene set test conducted and which methods are best suited to effectively prioritize gene sets with high phenotype relevance.
Availability: http://bioconductor.org/packages/GSEABenchmarkeR.
Contact: ludwig.geistlinger@sph.cuny.edu.
Keywords: RNA-seq; gene expression data; gene set analysis; microarray; pathway analysis.
© The Author(s) 2020. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures