

Transmission dynamics and evolutionary history of 2019-nCoV
- PMID: 32027035
- PMCID: PMC7166881
- DOI: 10.1002/jmv.25701
Transmission dynamics and evolutionary history of 2019-nCoV
Abstract
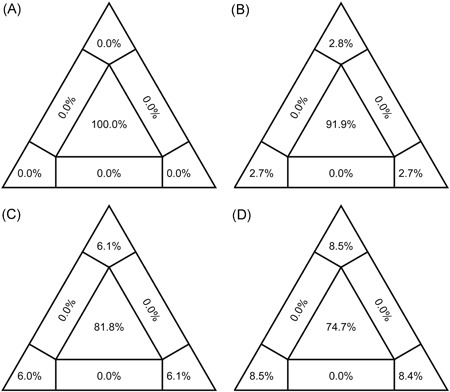
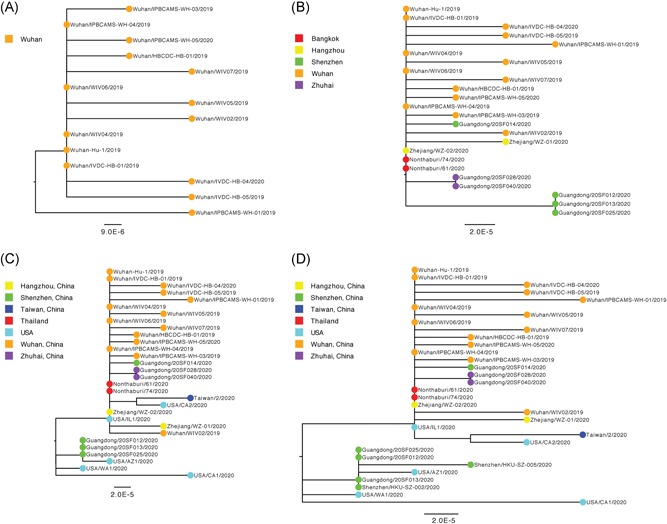
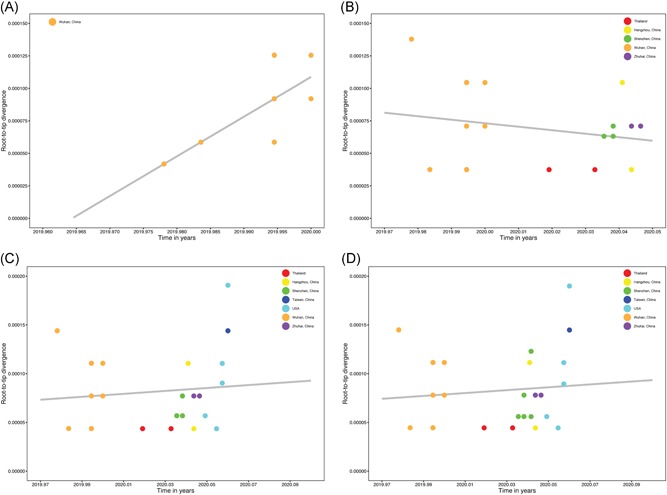
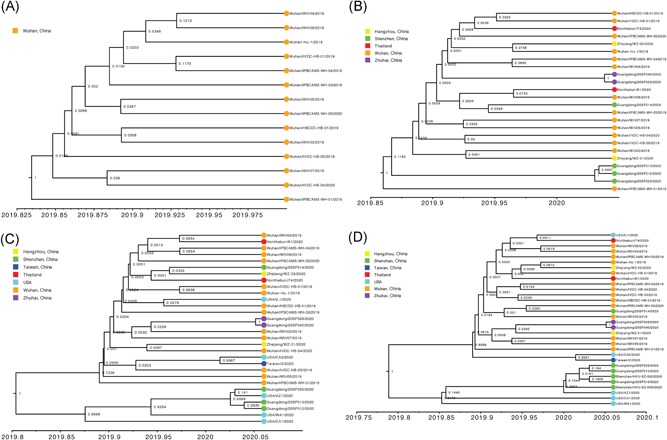
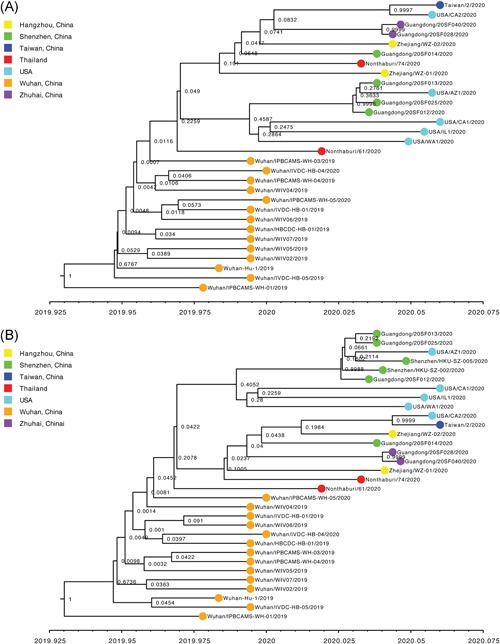
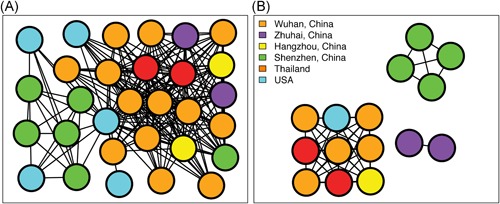
To investigate the time origin, genetic diversity, and transmission dynamics of the recent 2019-nCoV outbreak in China and beyond, a total of 32 genomes of virus strains sampled from China, Thailand, and the USA with sampling dates between 24 December 2019 and 23 January 2020 were analyzed. Phylogenetic, transmission network, and likelihood-mapping analyses of the genome sequences were performed. On the basis of the likelihood-mapping analysis, the increasing tree-like signals (from 0% to 8.2%, 18.2%, and 25.4%) over time may be indicative of increasing genetic diversity of 2019-nCoV in human hosts. We identified three phylogenetic clusters using the Bayesian inference framework and three transmission clusters using transmission network analysis, with only one cluster identified by both methods using the above genome sequences of 2019-nCoV strains. The estimated mean evolutionary rate for 2019-nCoV ranged from 1.7926 × 10-3 to 1.8266 × 10-3 substitutions per site per year. On the basis of our study, undertaking epidemiological investigations and genomic data surveillance could positively impact public health in terms of guiding prevention efforts to reduce 2019-nCOV transmission in real-time.
Keywords: 2019-nCoV; TMRCA; evolutionary rate; phylogenetic cluster; time to most recent common ancestor; transmission cluster.
© 2020 Wiley Periodicals, Inc.
Conflict of interest statement
The authors declare that there are no conflict of interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
