

Epigenetic driver mutations in ARID1A shape cancer immune phenotype and immunotherapy
- PMID: 32027624
- PMCID: PMC7190935
- DOI: 10.1172/JCI134402
Epigenetic driver mutations in ARID1A shape cancer immune phenotype and immunotherapy
Abstract
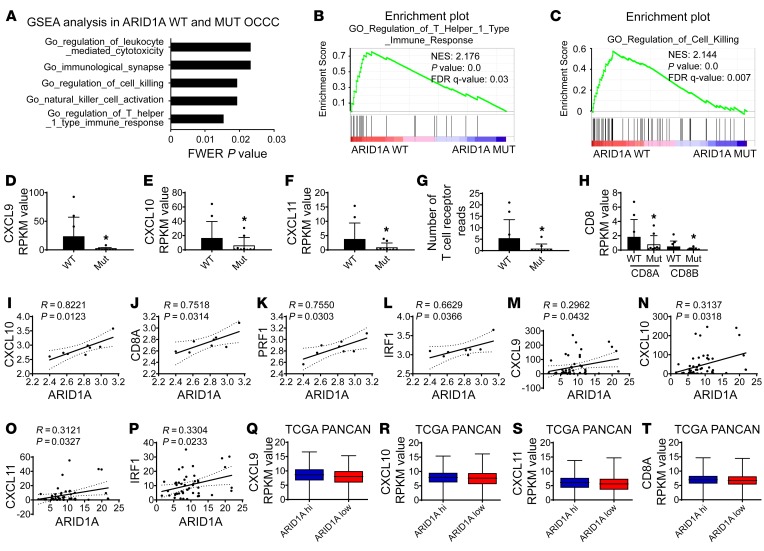
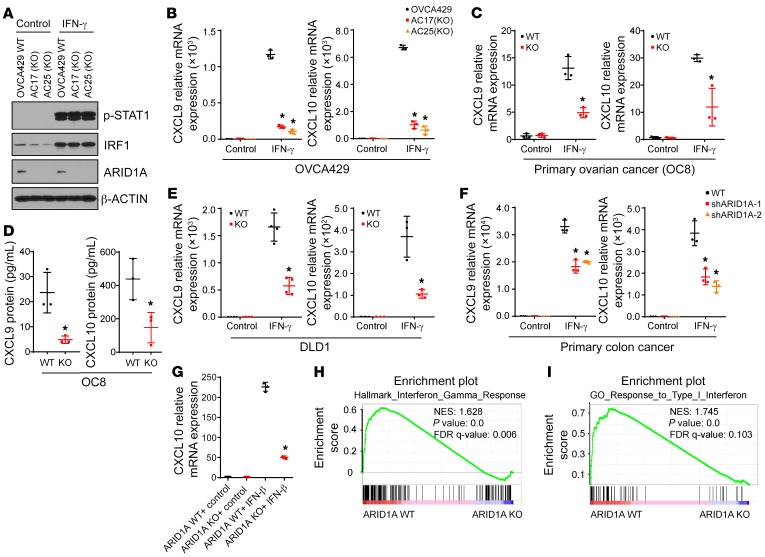
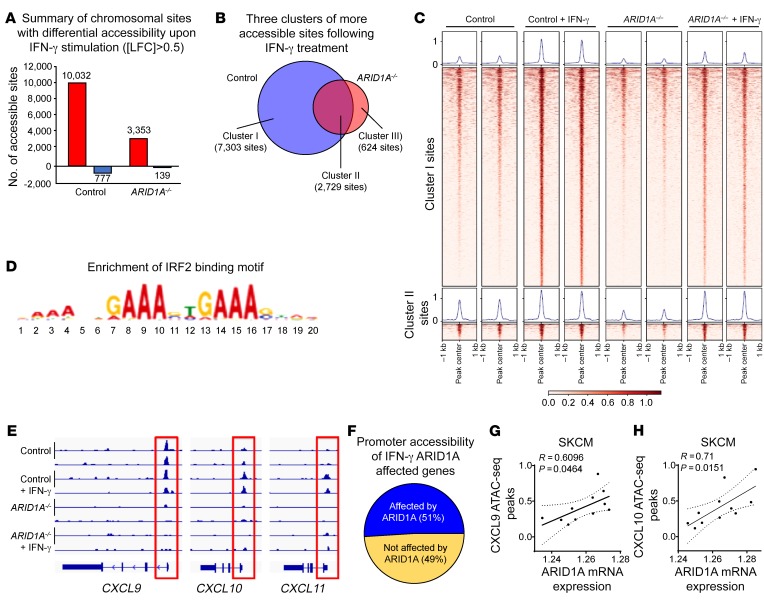
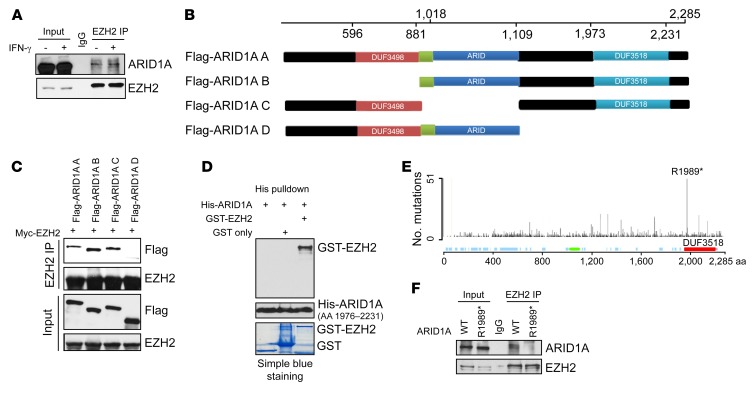
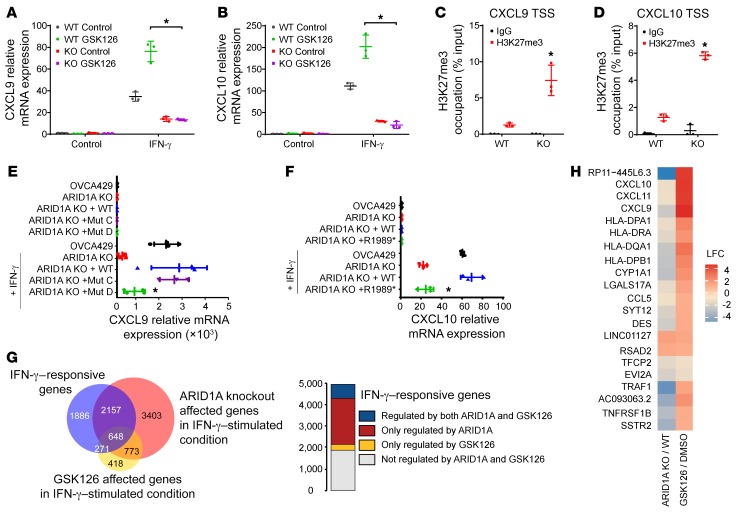
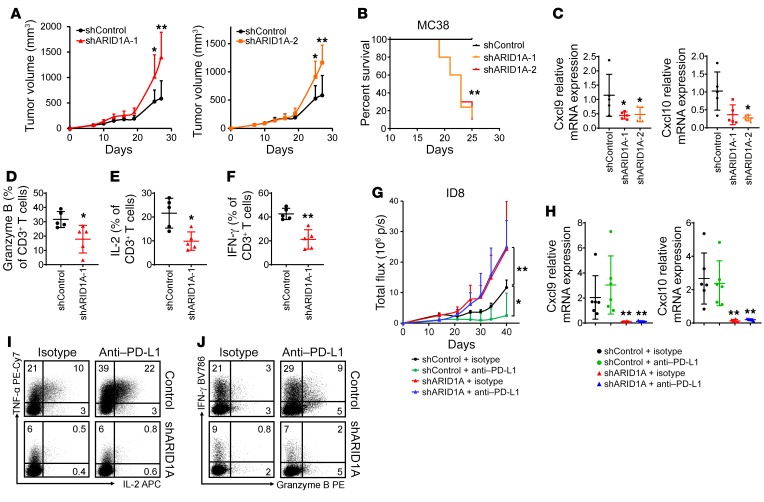
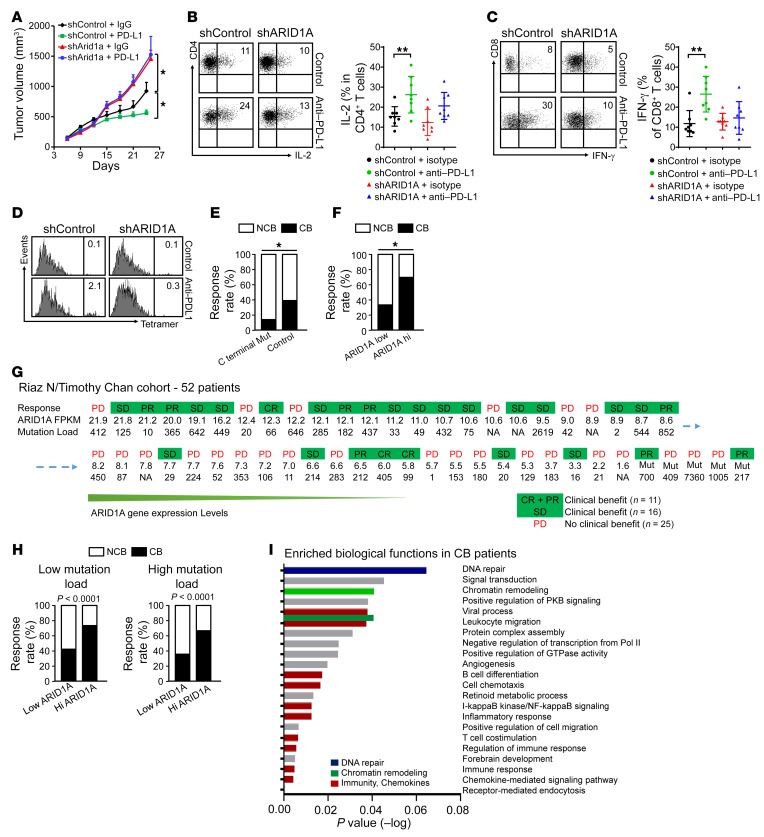
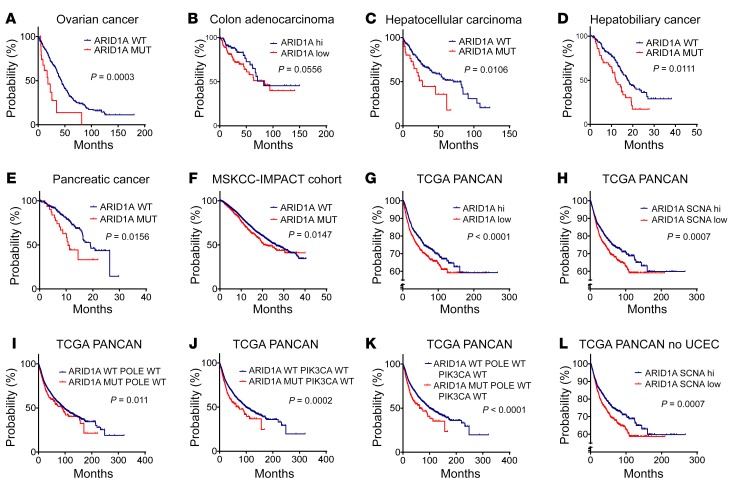
Whether mutations in cancer driver genes directly affect cancer immune phenotype and T cell immunity remains a standing question. ARID1A is a core member of the polymorphic BRG/BRM-associated factor chromatin remodeling complex. ARID1A mutations occur in human cancers and drive cancer development. Here, we studied the molecular, cellular, and clinical impact of ARID1A aberrations on cancer immunity. We demonstrated that ARID1A aberrations resulted in limited chromatin accessibility to IFN-responsive genes, impaired IFN gene expression, anemic T cell tumor infiltration, poor tumor immunity, and shortened host survival in many human cancer histologies and in murine cancer models. Impaired IFN signaling was associated with poor immunotherapy response. Mechanistically, ARID1A interacted with EZH2 via its carboxyl terminal and antagonized EZH2-mediated IFN responsiveness. Thus, the interaction between ARID1A and EZH2 defines cancer IFN responsiveness and immune evasion. Our work indicates that cancer epigenetic driver mutations can shape cancer immune phenotype and immunotherapy.
Keywords: Cancer immunotherapy; Immunology; T cells.
Conflict of interest statement
Figures
References
-
- Shin DS, et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. 2017;7(2):188–201. doi: 10.1158/2159-8290.CD-16-1223. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 CA193136/CA/NCI NIH HHS/United States
- P30 CA046592/CA/NCI NIH HHS/United States
- P30 CA010815/CA/NCI NIH HHS/United States
- R01 CA217510/CA/NCI NIH HHS/United States
- R01 CA152470/CA/NCI NIH HHS/United States
- P30 CA008748/CA/NCI NIH HHS/United States
- R35 CA232097/CA/NCI NIH HHS/United States
- R01 CA211016/CA/NCI NIH HHS/United States
- P50 CA186786/CA/NCI NIH HHS/United States
- R01 CA123088/CA/NCI NIH HHS/United States
- T32 AI007413/AI/NIAID NIH HHS/United States
- R01 CA099985/CA/NCI NIH HHS/United States
- R01 CA190176/CA/NCI NIH HHS/United States
- R01 CA214911/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
