

Extended Dynamically Weighted CASPT2: The Best of Two Worlds
- PMID: 32027802
- PMCID: PMC7307887
- DOI: 10.1021/acs.jctc.9b01129
Extended Dynamically Weighted CASPT2: The Best of Two Worlds
Abstract
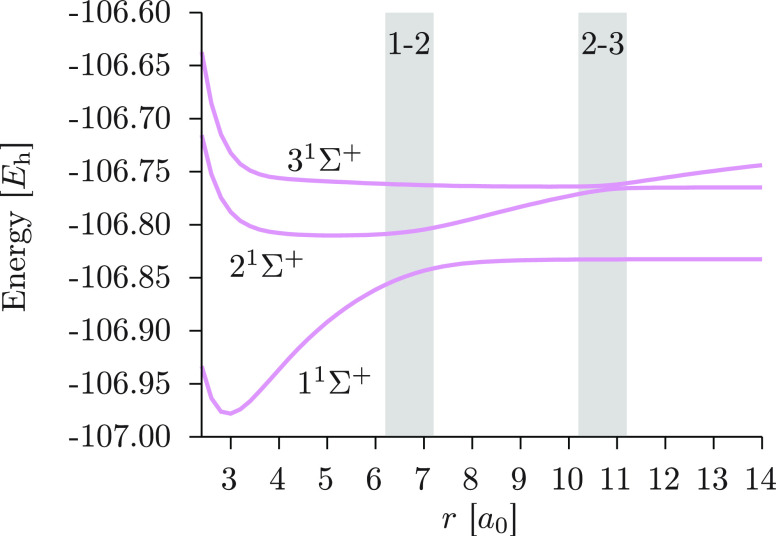
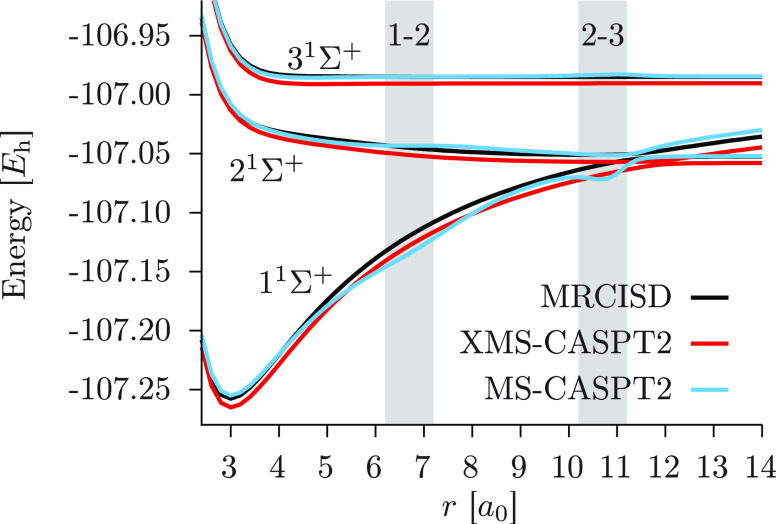
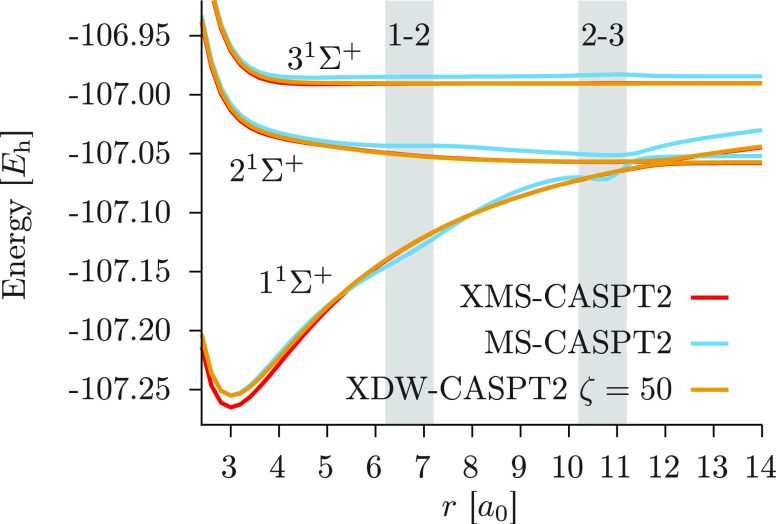
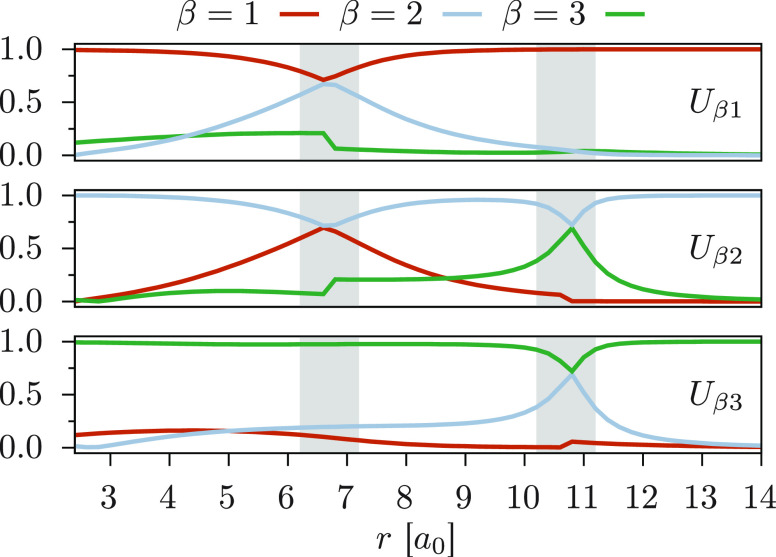
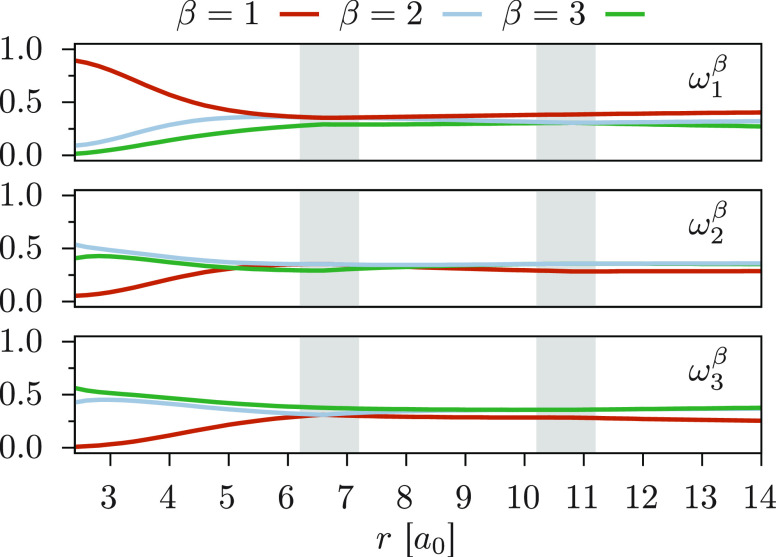
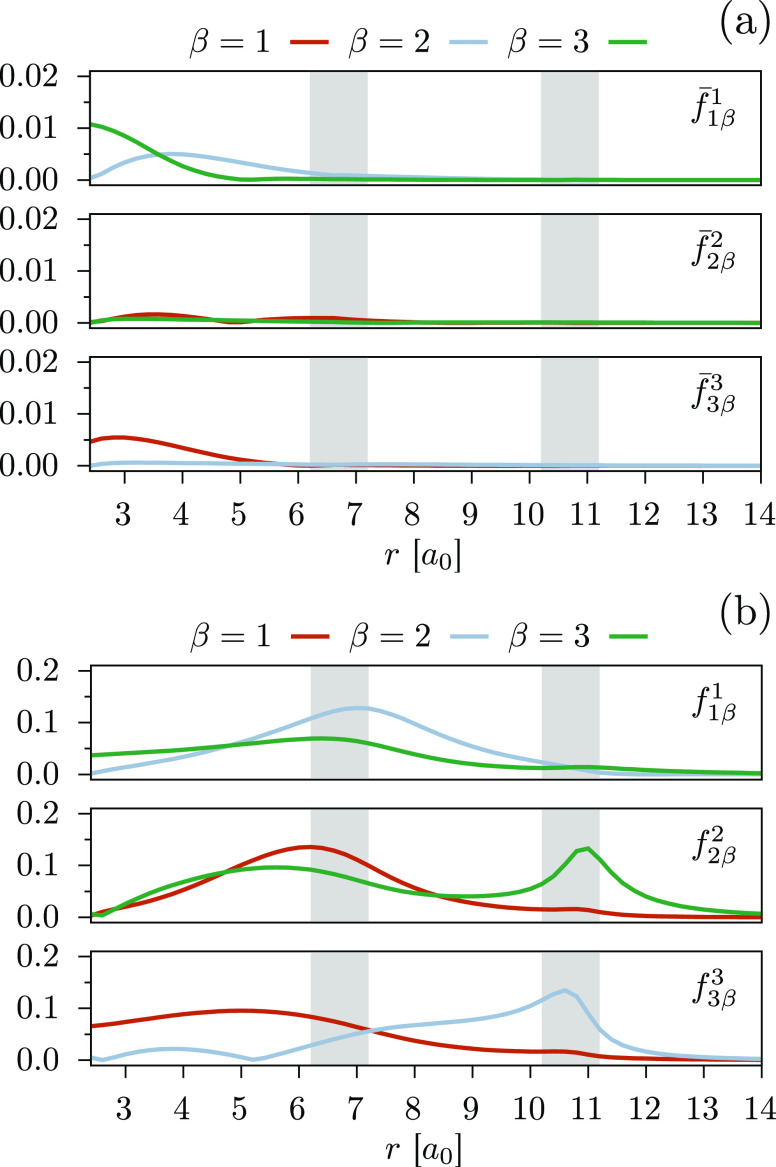
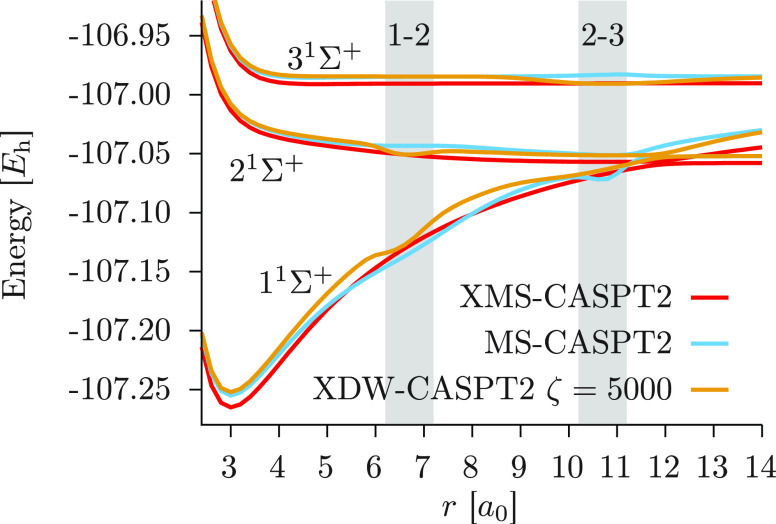
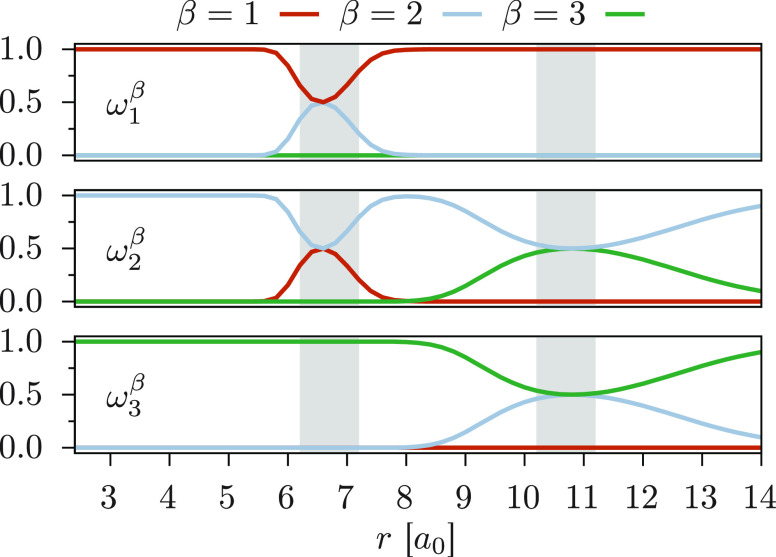
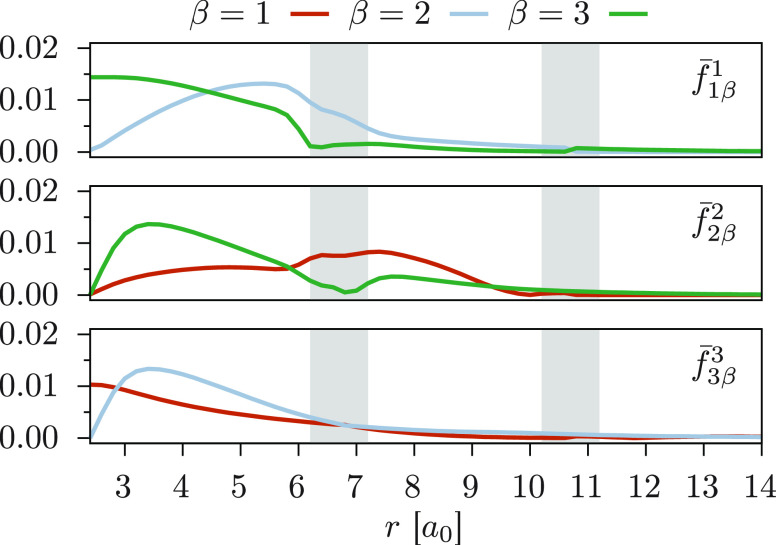
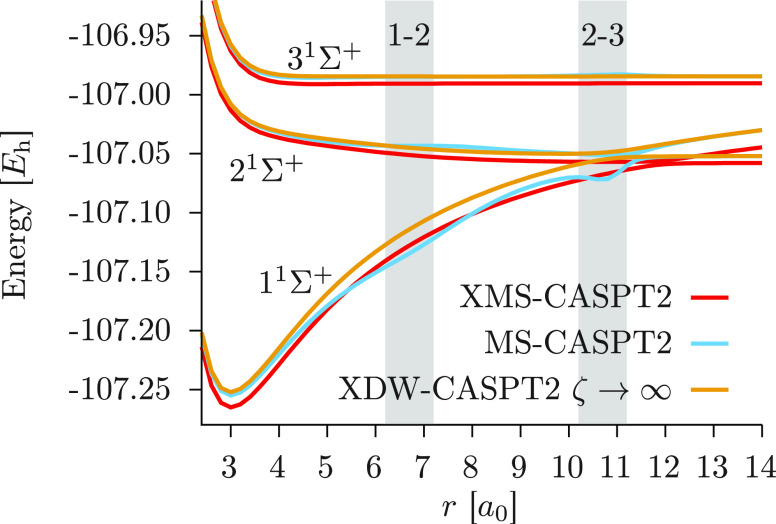
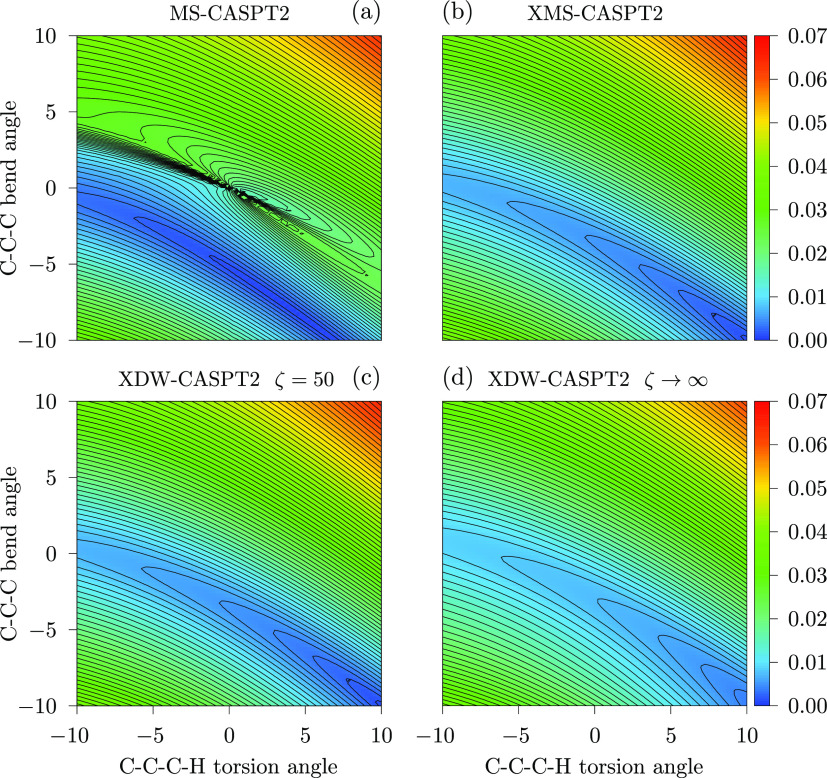
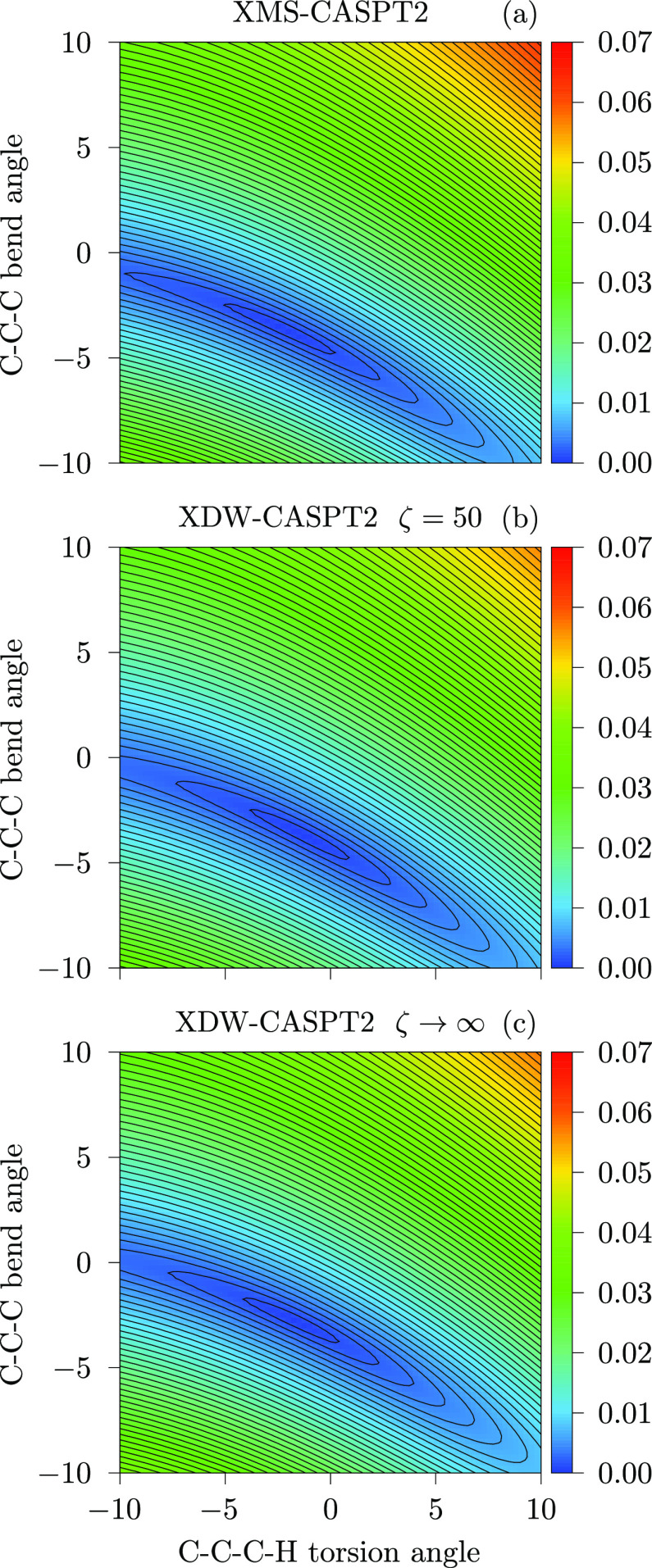
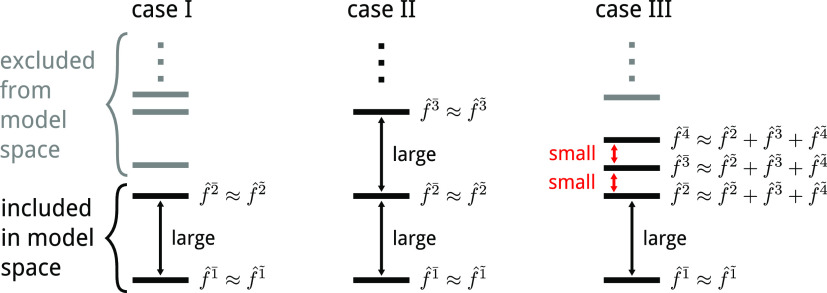
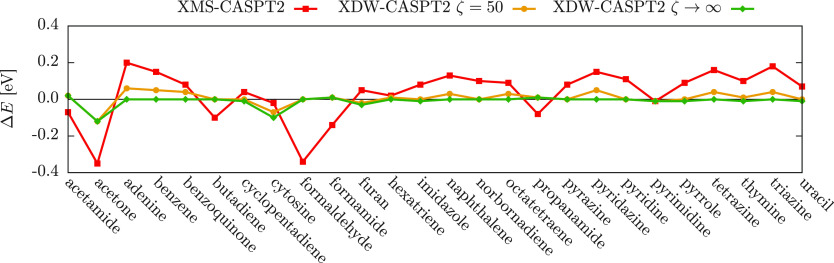
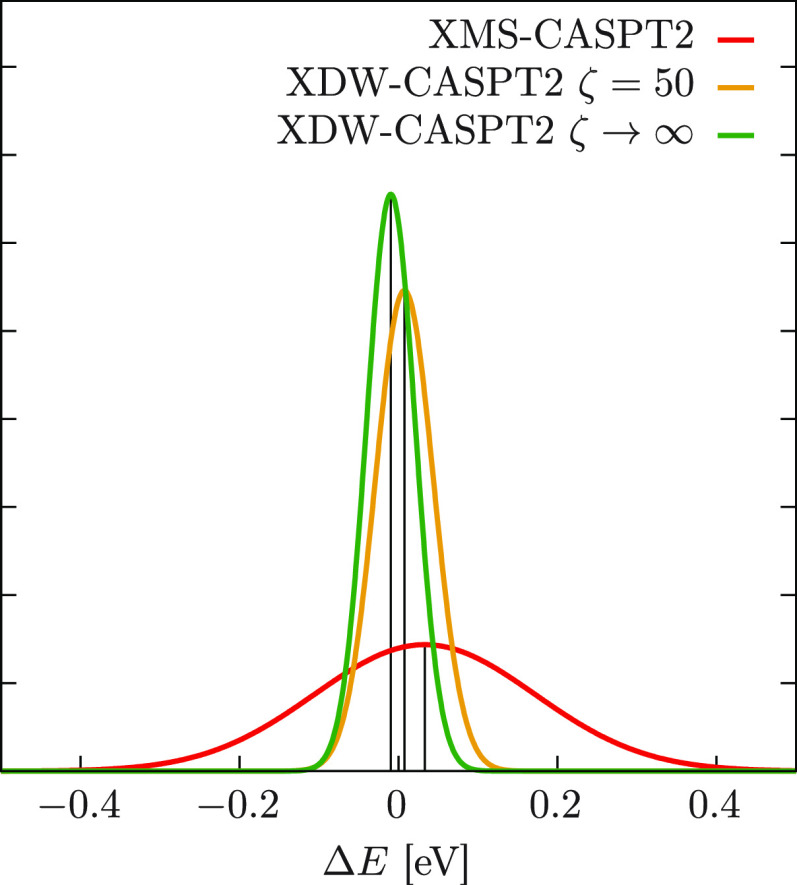
We introduce a new variant of the complete active space second-order perturbation theory (CASPT2) method that performs similarly to multistate CASPT2 (MS-CASPT2) in regions of the potential energy surface where the electronic states are energetically well separated and is akin to extended MS-CASPT2 (XMS-CASPT2) in case the underlying zeroth-order references are near-degenerate. Our approach follows a recipe analogous to that of XMS-CASPT2 to ensure approximate invariance under unitary transformations of the model states and a dynamic weighting scheme to smoothly interpolate the Fock operator between state-specific and state-average regimes. The resulting extended dynamically weighted CASPT2 (XDW-CASPT2) methodology possesses the most desirable features of both MS-CASPT2 and XMS-CASPT2, that is, the ability to provide accurate transition energies and correctly describe avoided crossings and conical intersections. The reliability of XDW-CASPT2 is assessed on a number of molecular systems. First, we consider the dissociation of lithium fluoride, highlighting the distinctive characteristics of the new approach. Second, the invariance of the theory is investigated by studying the conical intersection of the distorted allene molecule. Finally, the relative accuracy in the calculation of vertical excitation energies is benchmarked on a set of 26 organic compounds. We found that XDW-CASPT2, albeit being only approximately invariant, produces smooth potential energy surfaces around conical intersections and avoided crossings, performing equally well to the strictly invariant XMS-CASPT2 method. The accuracy of vertical transition energies is almost identical to MS-CASPT2, with a mean absolute deviation of 0.01-0.02 eV, in contrast to 0.12 eV for XMS-CASPT2.
Conflict of interest statement
The authors declare no competing financial interest.
Figures

References
LinkOut - more resources
Full Text Sources
