

Itaconate modulates tricarboxylic acid and redox metabolism to mitigate reperfusion injury
- PMID: 32029222
- PMCID: PMC6961711
- DOI: 10.1016/j.molmet.2019.11.019
Itaconate modulates tricarboxylic acid and redox metabolism to mitigate reperfusion injury
Abstract
Objectives: Cerebral ischemia/reperfusion (IR) drives oxidative stress and injurious metabolic processes that lead to redox imbalance, inflammation, and tissue damage. However, the key mediators of reperfusion injury remain unclear, and therefore, there is considerable interest in therapeutically targeting metabolism and the cellular response to oxidative stress.
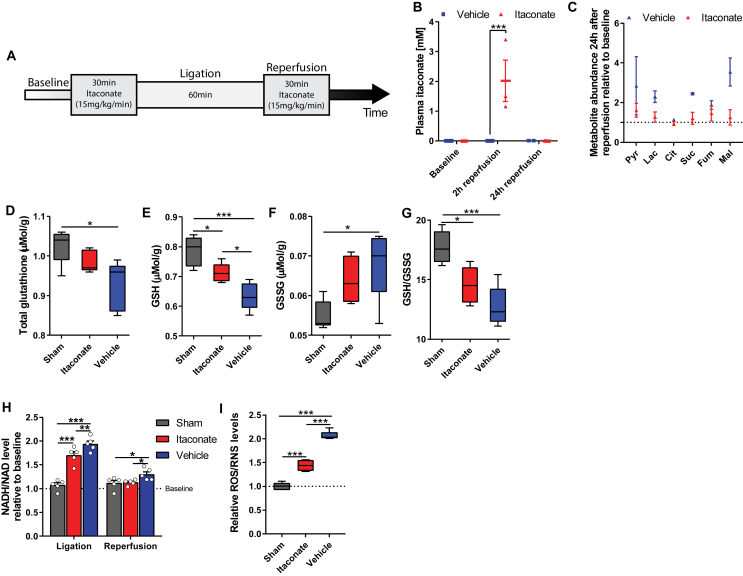
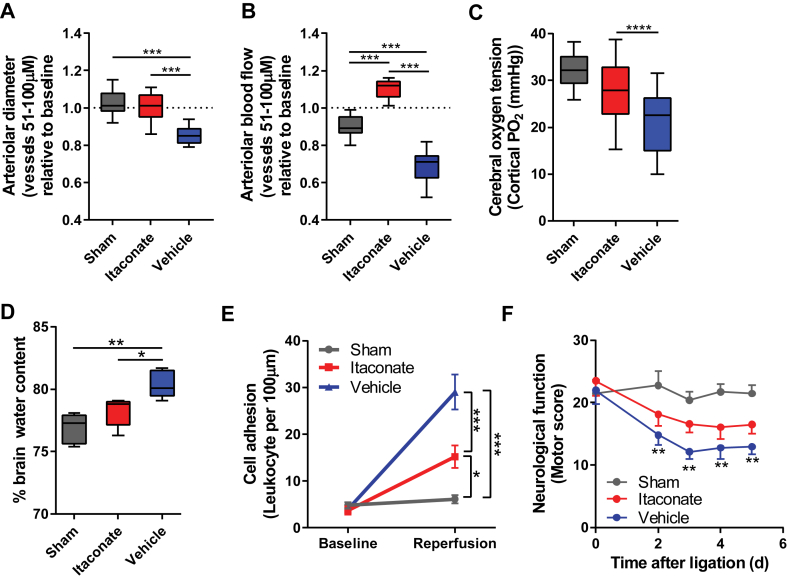
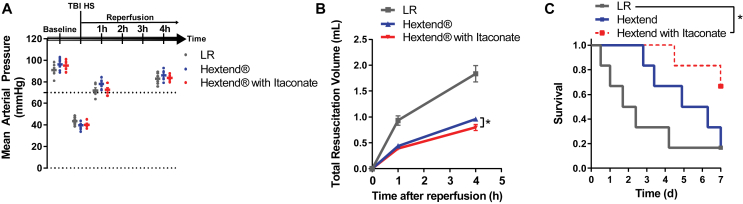
Methods: The objective of this study was to investigate the molecular, metabolic, and physiological impact of itaconate treatment to mitigate reperfusion injuries in in vitro and in vivo model systems. We conducted metabolic flux and bioenergetic studies in response to exogenous itaconate treatment in cultures of primary rat cortical neurons and astrocytes. In addition, we administered itaconate to mouse models of cerebral reperfusion injury with ischemia or traumatic brain injury followed by hemorrhagic shock resuscitation. We quantitatively characterized the metabolite levels, neurological behavior, markers of redox stress, leukocyte adhesion, arterial blood flow, and arteriolar diameter in the brains of the treated/untreated mice.
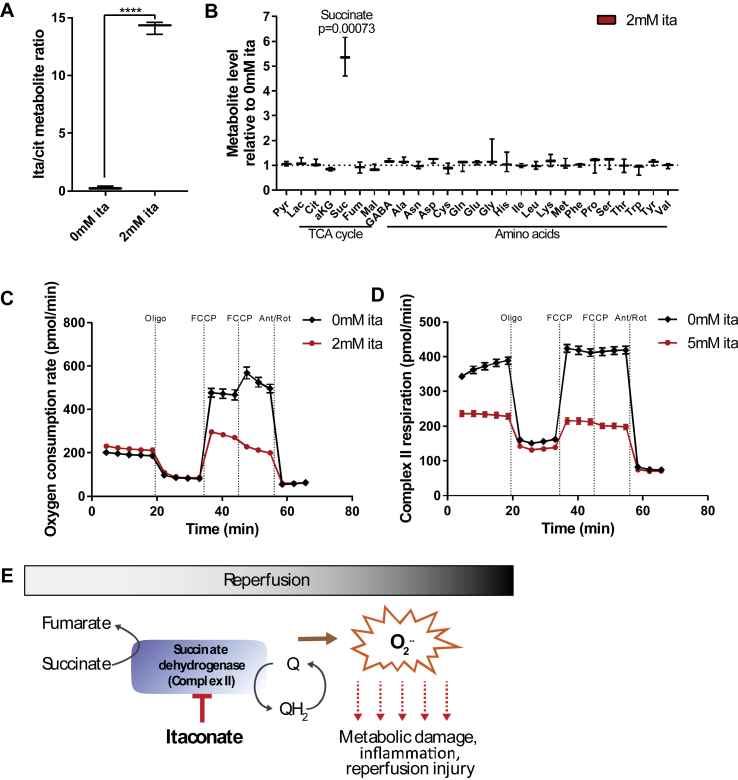
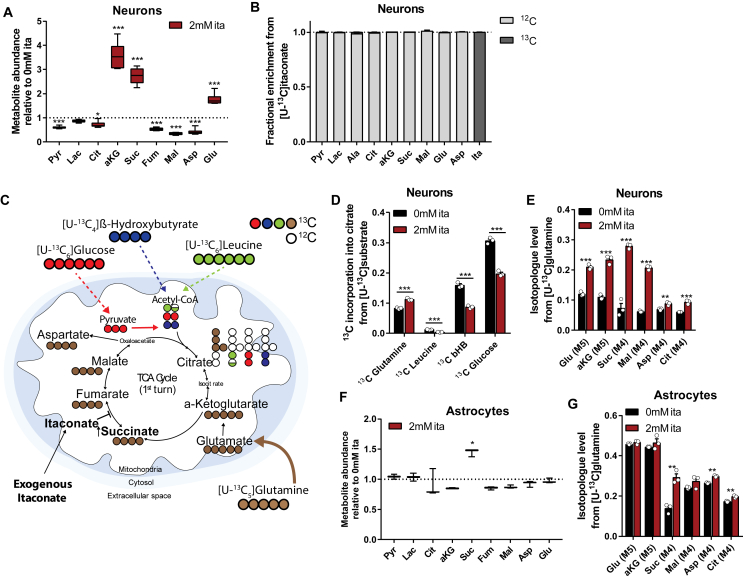
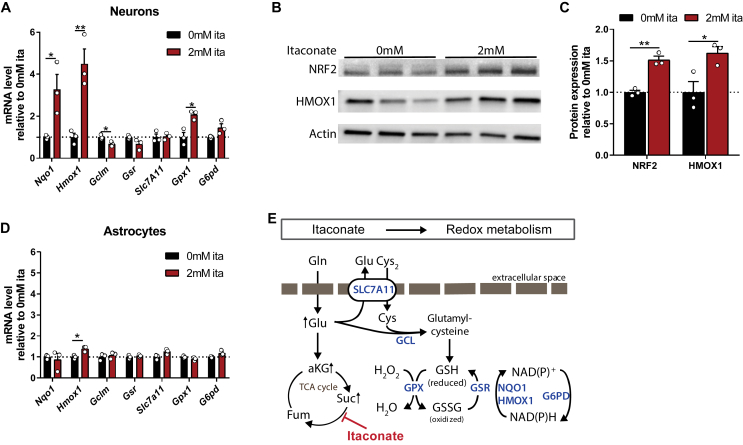
Results: We demonstrate that the "immunometabolite" itaconate slowed tricarboxylic acid (TCA) cycle metabolism and buffered redox imbalance via succinate dehydrogenase (SDH) inhibition and induction of anti-oxidative stress response in primary cultures of astrocytes and neurons. The addition of itaconate to reperfusion fluids after mouse cerebral IR injury increased glutathione levels and reduced reactive oxygen/nitrogen species (ROS/RNS) to improve neurological function. Plasma organic acids increased post-reperfusion injury, while administration of itaconate normalized these metabolites. In mouse cranial window models, itaconate significantly improved hemodynamics while reducing leukocyte adhesion. Further, itaconate supplementation increased survival in mice experiencing traumatic brain injury (TBI) and hemorrhagic shock.
Conclusions: We hypothesize that itaconate transiently inhibits SDH to gradually "awaken" mitochondrial function upon reperfusion that minimizes ROS and tissue damage. Collectively, our data indicate that itaconate acts as a mitochondrial regulator that controls redox metabolism to improve physiological outcomes associated with IR injury.
Keywords: Brain injury; Cerebral ischemia/reperfusion (IR); Itaconate; Mitochondrial metabolism; Redox stress; Succinate dehydrogenase (SDH).
Copyright © 2019 The Authors. Published by Elsevier GmbH.. All rights reserved.
Figures
References
-
- White B.C., Sullivan J.M., DeGracia D.J., O'Neil B.J., Neumar R.W., Grossman L.I. Brain ischemia and reperfusion: molecular mechanisms of neuronal injury. Journal of the Neurological Sciences. 2000;179(S 1–2):1–33. - PubMed
-
- McCord J.M. Oxygen-derived radicals: a link between reperfusion injury and inflammation. Federation Proceedings. 1987 - PubMed
-
- Chouchani E.T., Pell V.R., James A.M., Work L.M., Saeb-Parsy K., Frezza C. A unifying mechanism for mitochondrial superoxide production during ischemia-reperfusion injury. Cell Metabolism. 2016;23(2):254–263. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
