

PARP and PARG inhibitors in cancer treatment
- PMID: 32029455
- PMCID: PMC7050487
- DOI: 10.1101/gad.334516.119
PARP and PARG inhibitors in cancer treatment
Abstract
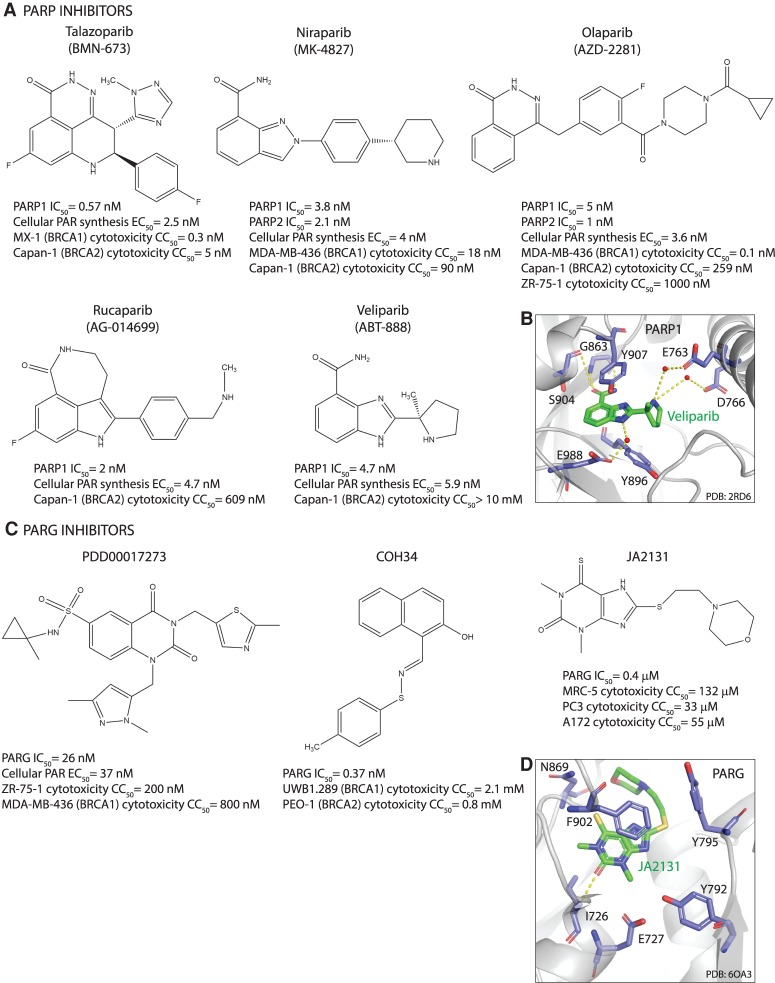
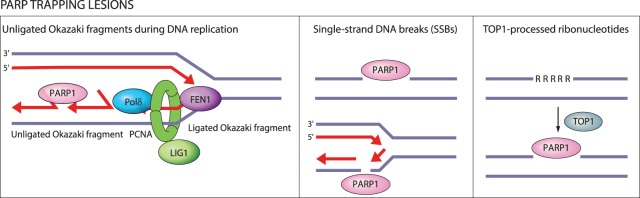
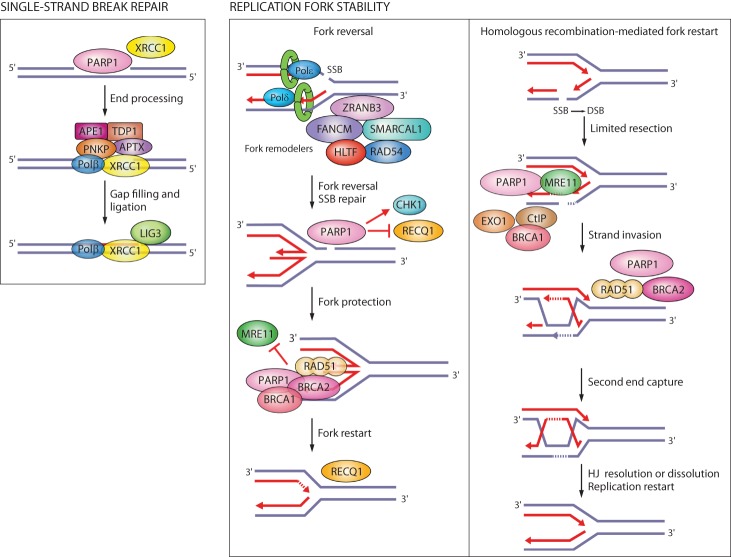
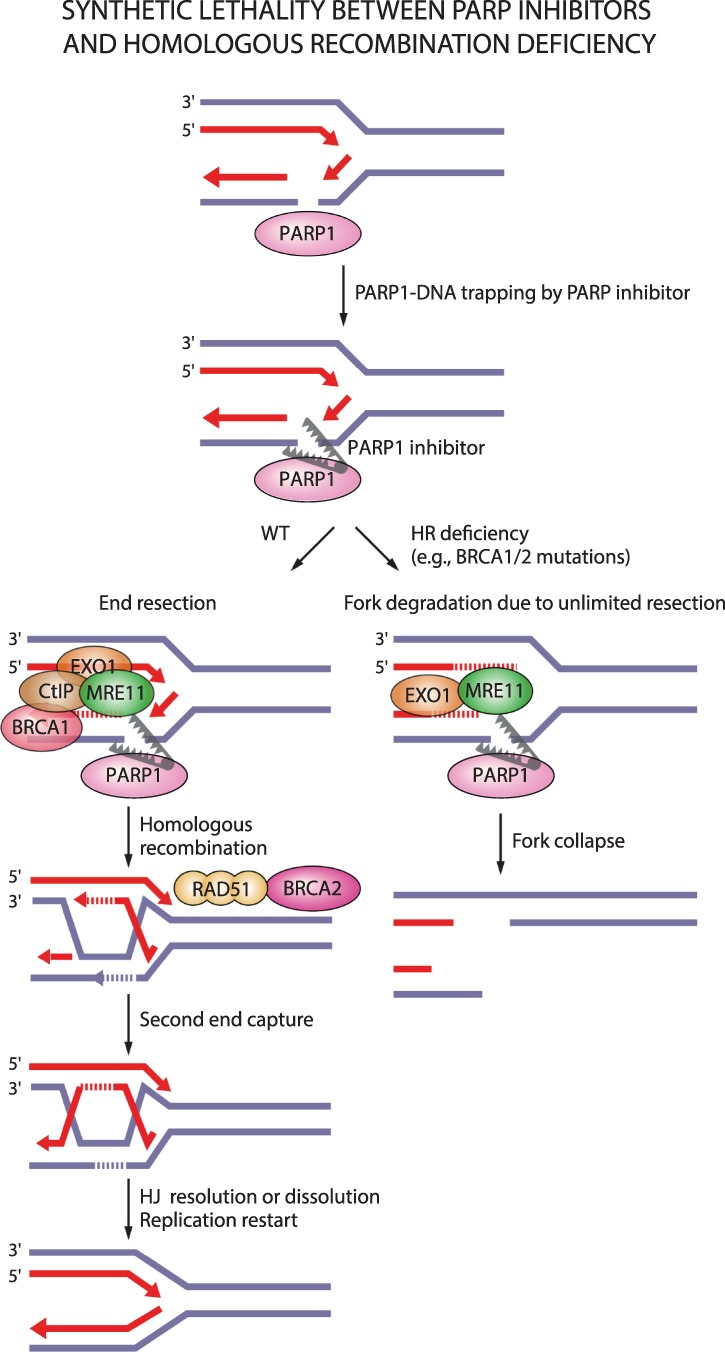
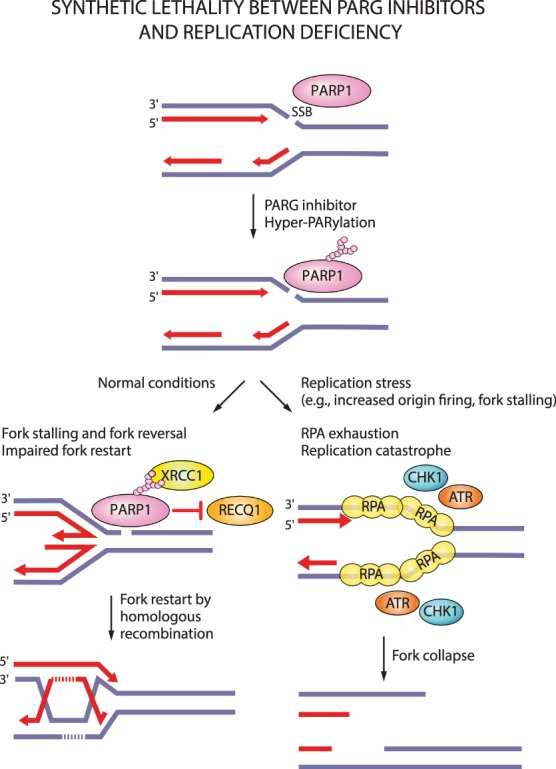
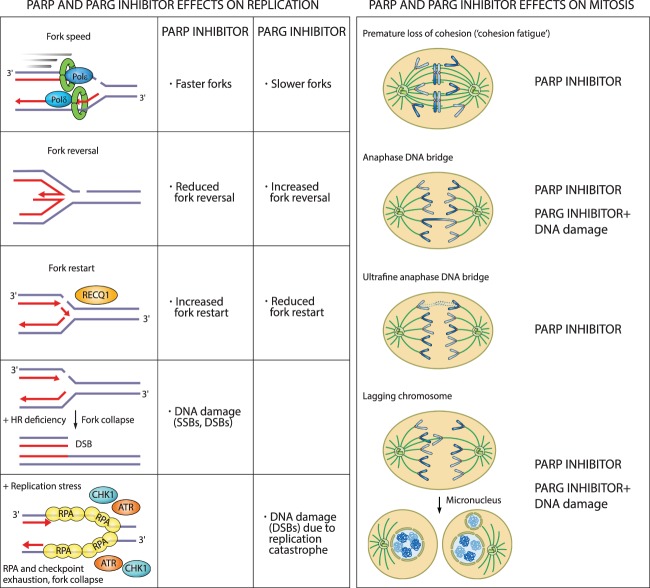
Oxidative and replication stress underlie genomic instability of cancer cells. Amplifying genomic instability through radiotherapy and chemotherapy has been a powerful but nonselective means of killing cancer cells. Precision medicine has revolutionized cancer therapy by putting forth the concept of selective targeting of cancer cells. Poly(ADP-ribose) polymerase (PARP) inhibitors represent a successful example of precision medicine as the first drugs targeting DNA damage response to have entered the clinic. PARP inhibitors act through synthetic lethality with mutations in DNA repair genes and were approved for the treatment of BRCA mutated ovarian and breast cancer. PARP inhibitors destabilize replication forks through PARP DNA entrapment and induce cell death through replication stress-induced mitotic catastrophe. Inhibitors of poly(ADP-ribose) glycohydrolase (PARG) exploit and exacerbate replication deficiencies of cancer cells and may complement PARP inhibitors in targeting a broad range of cancer types with different sources of genomic instability. Here I provide an overview of the molecular mechanisms and cellular consequences of PARP and PARG inhibition. I highlight clinical performance of four PARP inhibitors used in cancer therapy (olaparib, rucaparib, niraparib, and talazoparib) and discuss the predictive biomarkers of inhibitor sensitivity, mechanisms of resistance as well as the means of overcoming them through combination therapy.
Keywords: PARG inhibitor; PARP inhibitor; cancer therapy; poly(ADP-ribose) glycohydrolase; poly(ADP-ribose) polymerases.
© 2020 Slade; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Abkevich V, Timms KM, Hennessy BT, Potter J, Carey MS, Meyer LA, Smith-McCune K, Broaddus R, Lu KH, Chen J, et al. 2012. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br J Cancer 107: 1776–1782. 10.1038/bjc.2012.451 - DOI - PMC - PubMed
-
- Allison Stewart C, Tong P, Cardnell RJ, Sen T, Li L, Gay CM, Masrorpour F, Fan Y, Bara RO, Feng Y, et al. 2017. Dynamic variations in epithelial-to-mesenchymal transition (EMT), ATM, and SLFN11 govern response to PARP inhibitors and cisplatin in small cell lung cancer. Oncotarget 8: 28575–28587. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical