

Interferon-β Plays a Detrimental Role in Experimental Traumatic Brain Injury by Enhancing Neuroinflammation That Drives Chronic Neurodegeneration
- PMID: 32029532
- PMCID: PMC7083281
- DOI: 10.1523/JNEUROSCI.2516-19.2020
Interferon-β Plays a Detrimental Role in Experimental Traumatic Brain Injury by Enhancing Neuroinflammation That Drives Chronic Neurodegeneration
Abstract
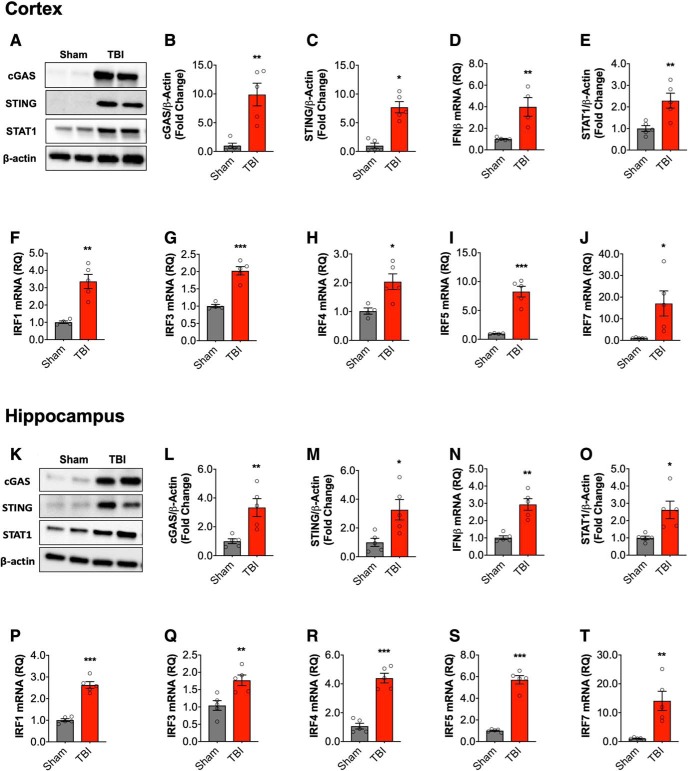
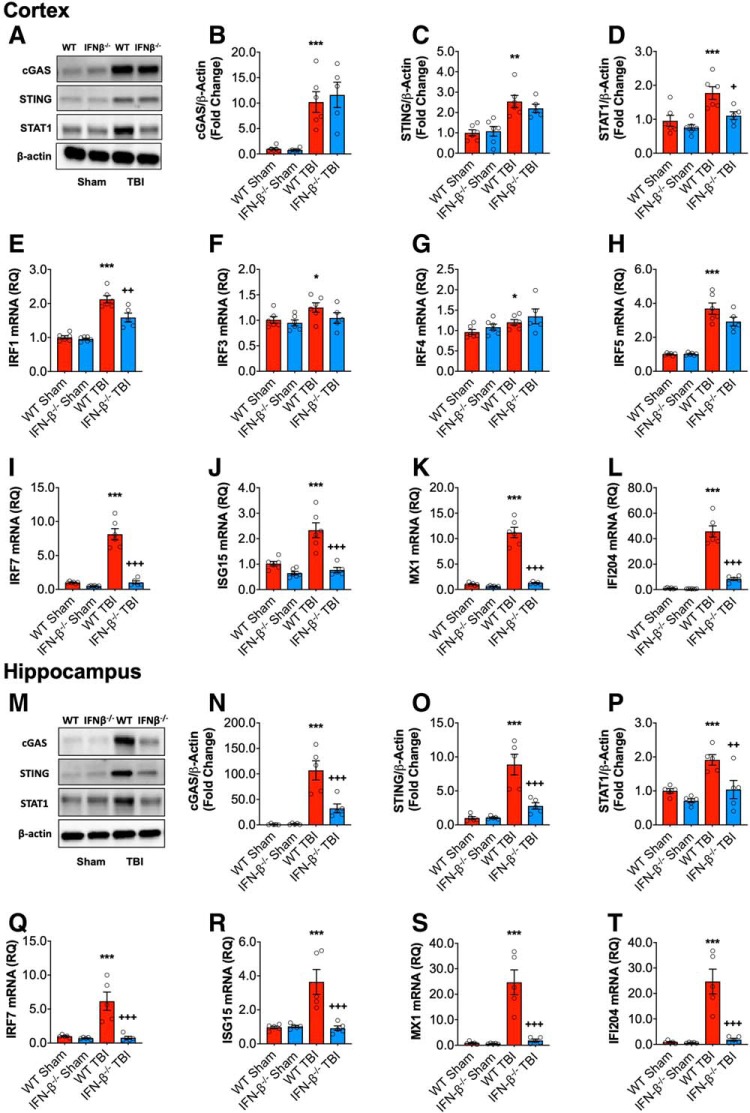
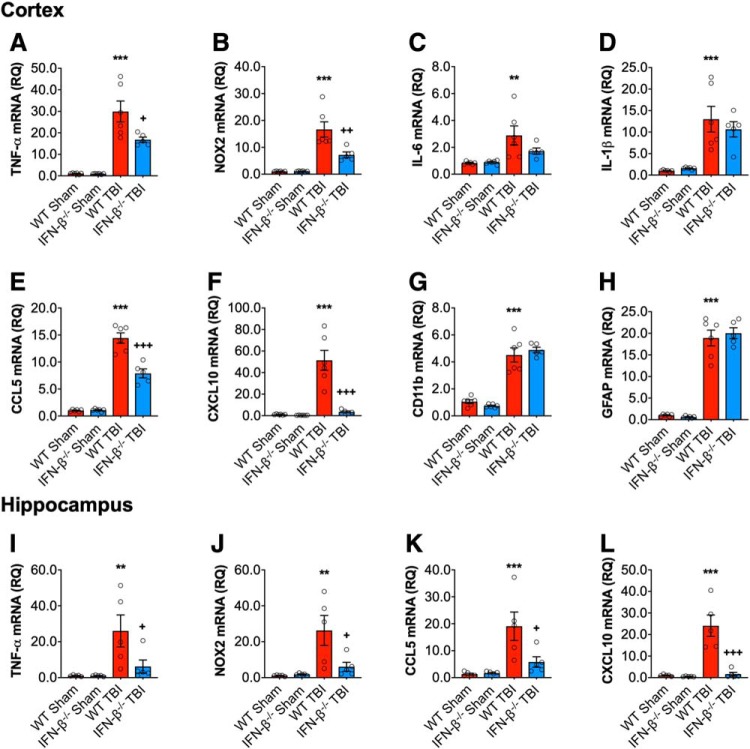
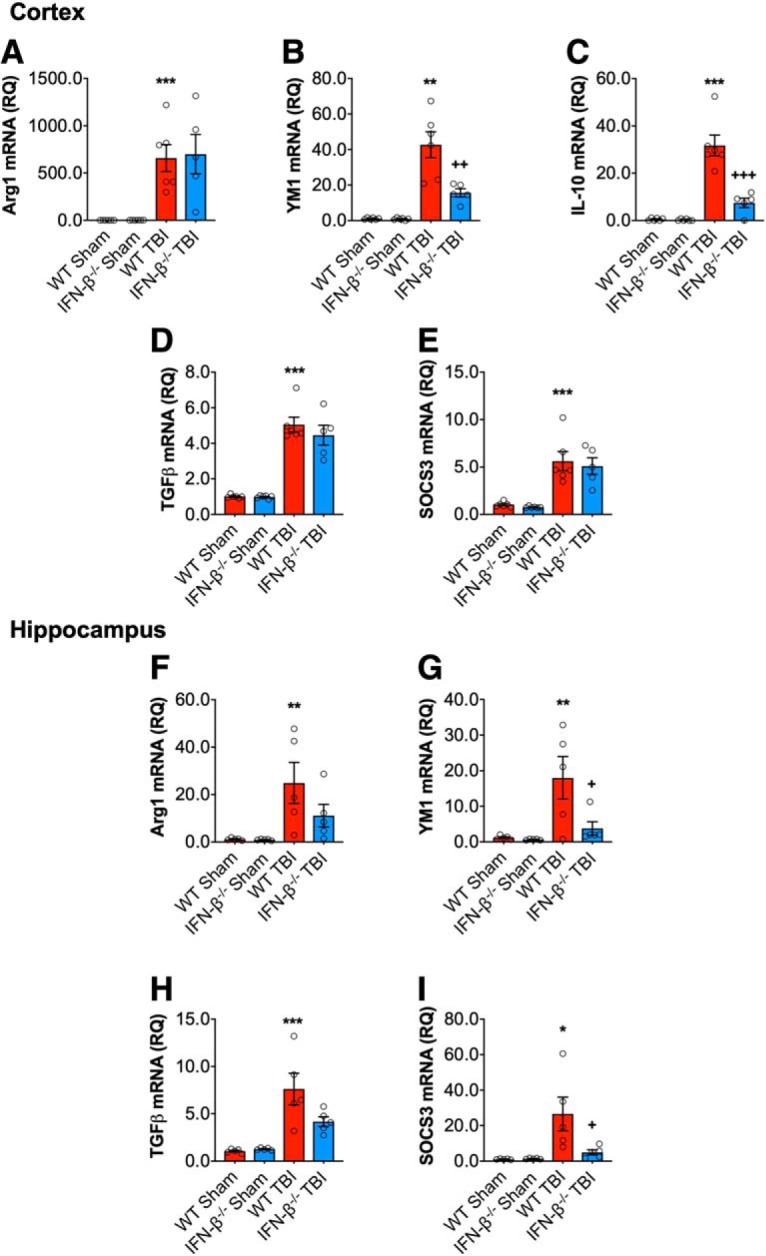
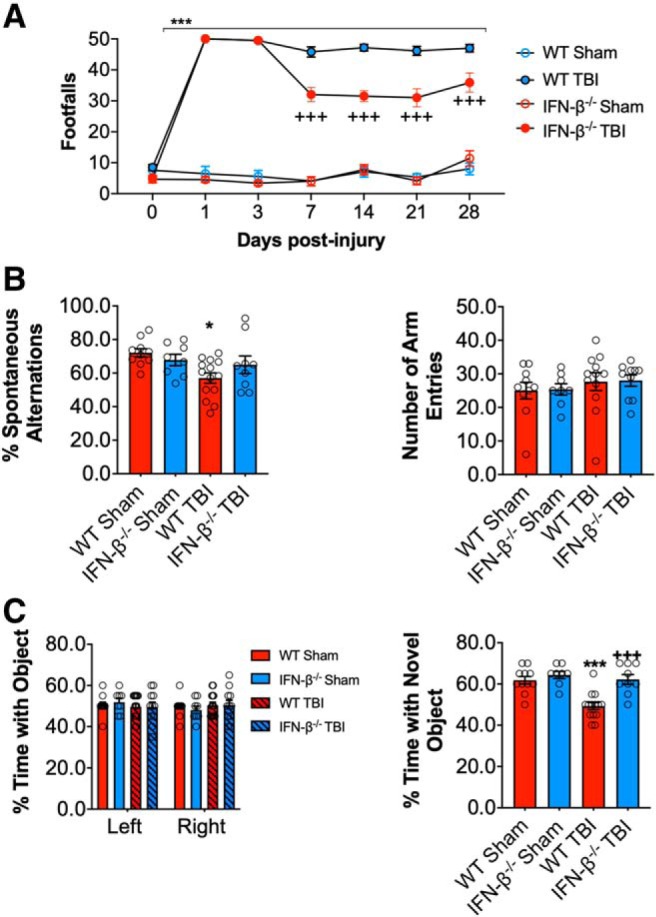
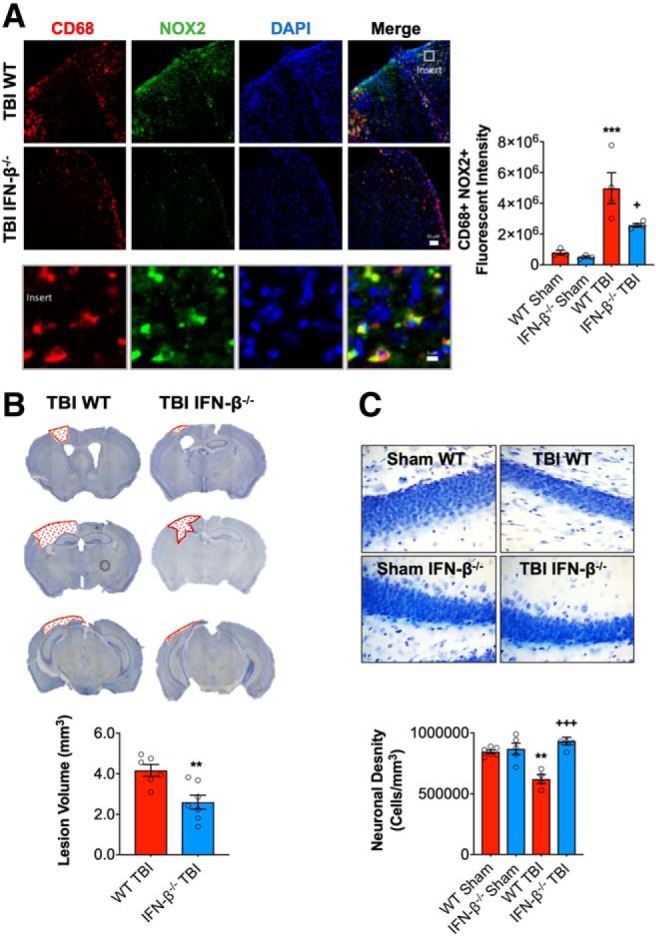
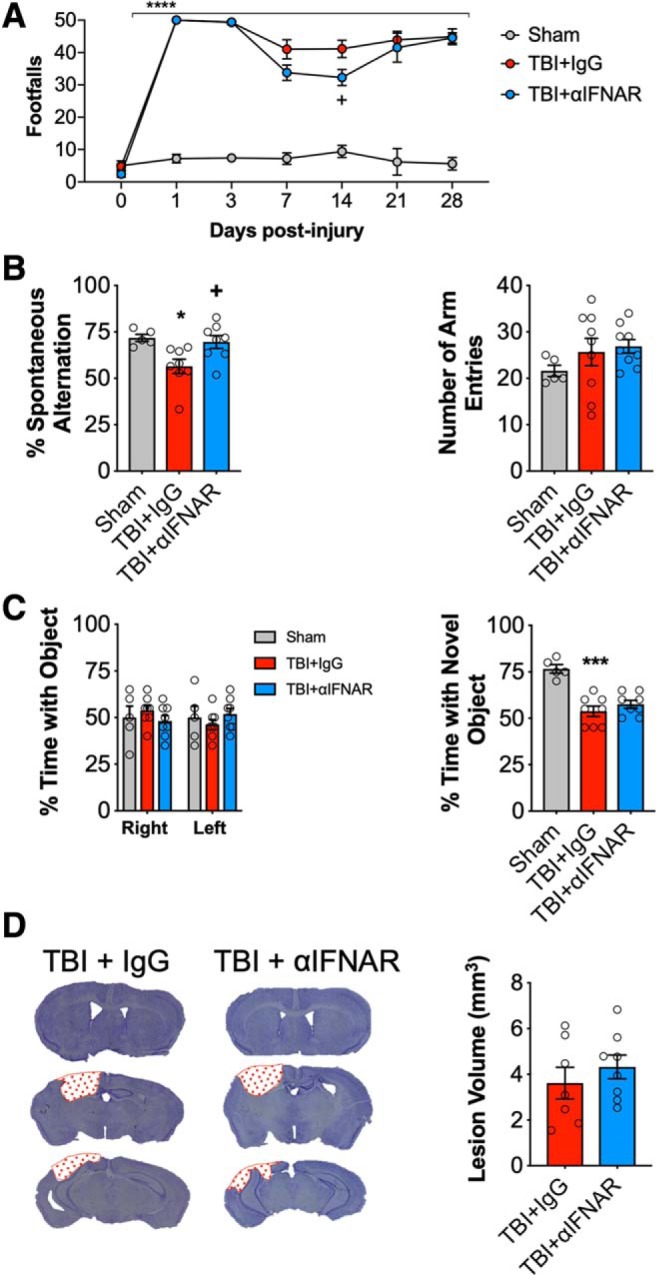
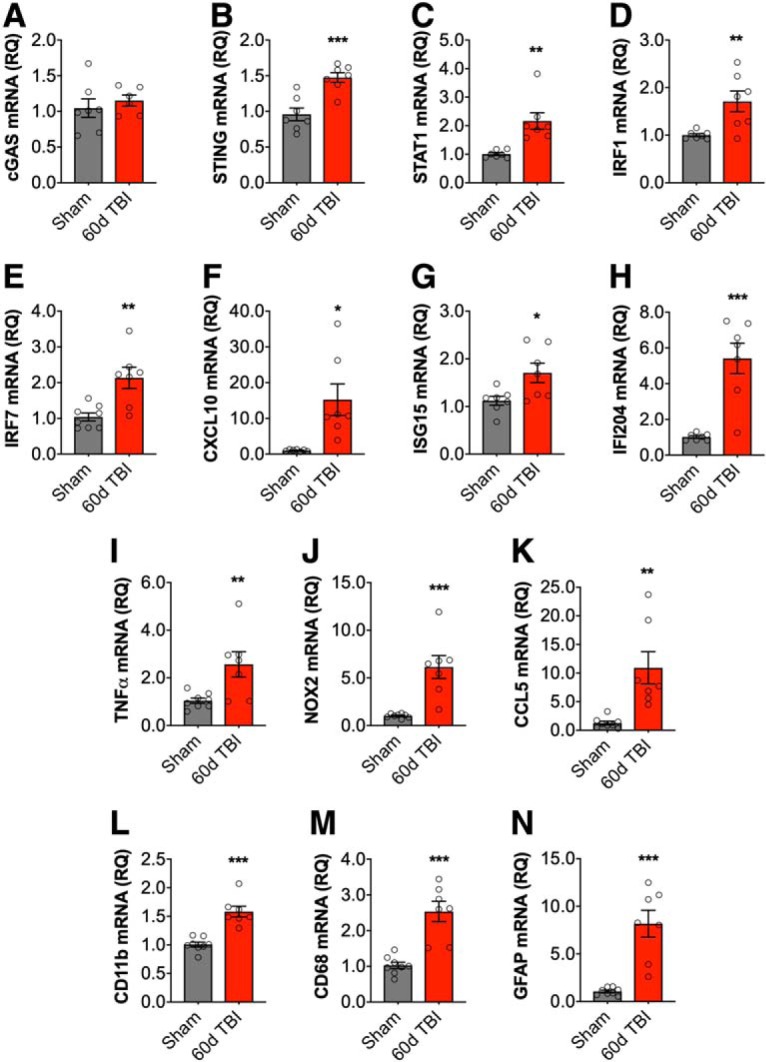
DNA damage and type I interferons (IFNs) contribute to inflammatory responses after traumatic brain injury (TBI). TBI-induced activation of microglia and peripherally-derived inflammatory macrophages may lead to tissue damage and neurological deficits. Here, we investigated the role of IFN-β in secondary injury after TBI using a controlled cortical impact model in adult male IFN-β-deficient (IFN-β-/-) mice and assessed post-traumatic neuroinflammatory responses, neuropathology, and long-term functional recovery. TBI increased expression of DNA sensors cyclic GMP-AMP synthase and stimulator of interferon genes in wild-type (WT) mice. IFN-β and other IFN-related and neuroinflammatory genes were also upregulated early and persistently after TBI. TBI increased expression of proinflammatory mediators in the cortex and hippocampus of WT mice, whereas levels were mitigated in IFN-β-/- mice. Moreover, long-term microglia activation, motor, and cognitive function impairments were decreased in IFN-β-/- TBI mice compared with their injured WT counterparts; improved neurological recovery was associated with reduced lesion volume and hippocampal neurodegeneration in IFN-β-/- mice. Continuous central administration of a neutralizing antibody to the IFN-α/β receptor (IFNAR) for 3 d, beginning 30 min post-injury, reversed early cognitive impairments in TBI mice and led to transient improvements in motor function. However, anti-IFNAR treatment did not improve long-term functional recovery or decrease TBI neuropathology at 28 d post-injury. In summary, TBI induces a robust neuroinflammatory response that is associated with increased expression of IFN-β and other IFN-related genes. Inhibition of IFN-β reduces post-traumatic neuroinflammation and neurodegeneration, resulting in improved neurological recovery. Thus, IFN-β may be a potential therapeutic target for TBI.SIGNIFICANCE STATEMENT TBI frequently causes long-term neurological and psychiatric changes in head injury patients. TBI-induced secondary injury processes including persistent neuroinflammation evolve over time and can contribute to chronic neurological impairments. The present study demonstrates that TBI is followed by robust activation of type I IFN pathways, which have been implicated in microglial-associated neuroinflammation and chronic neurodegeneration. We examined the effects of genetic or pharmacological inhibition of IFN-β, a key component of type I IFN mechanisms to address its role in TBI pathophysiology. Inhibition of IFN-β signaling resulted in reduced neuroinflammation, attenuated neurobehavioral deficits, and limited tissue loss long after TBI. These preclinical findings suggest that IFN-β may be a potential therapeutic target for TBI.
Keywords: interferon-β; neurodegeneration; neuroinflammation; neuroprotection; traumatic brain injury; type I interferons.
Copyright © 2020 the authors.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials