

Down syndrome
- PMID: 32029743
- PMCID: PMC8428796
- DOI: 10.1038/s41572-019-0143-7
Down syndrome
Abstract
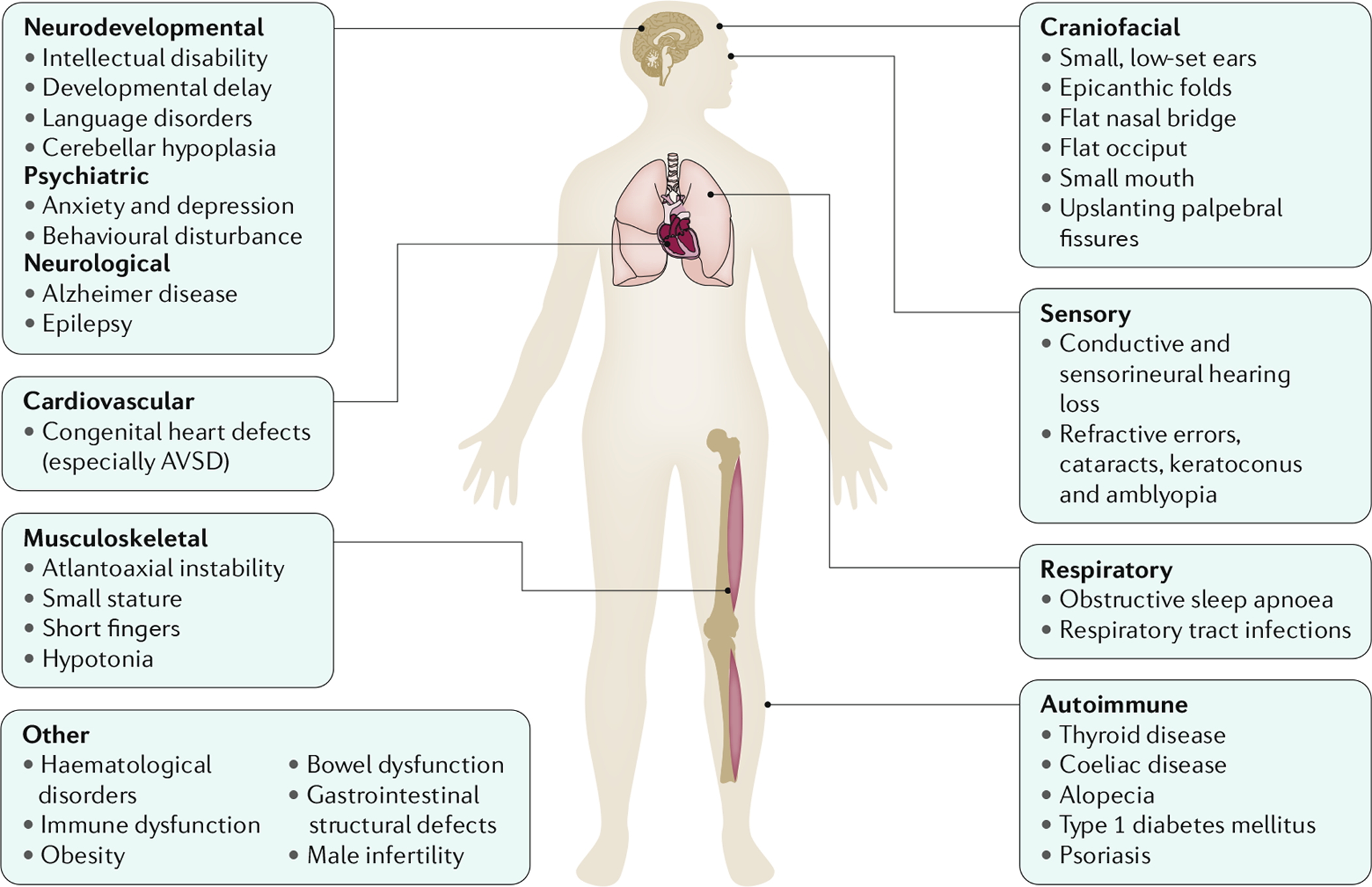
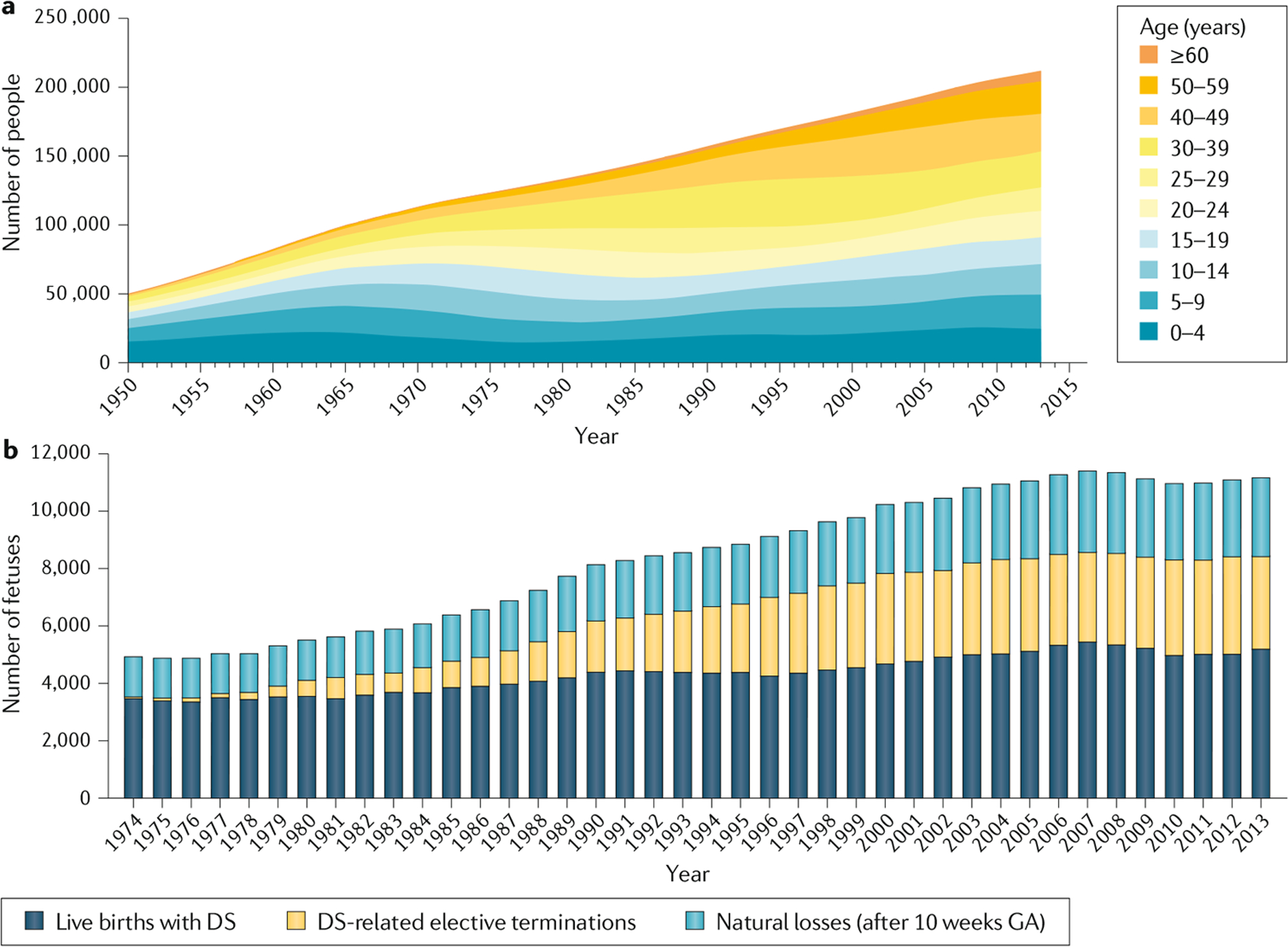
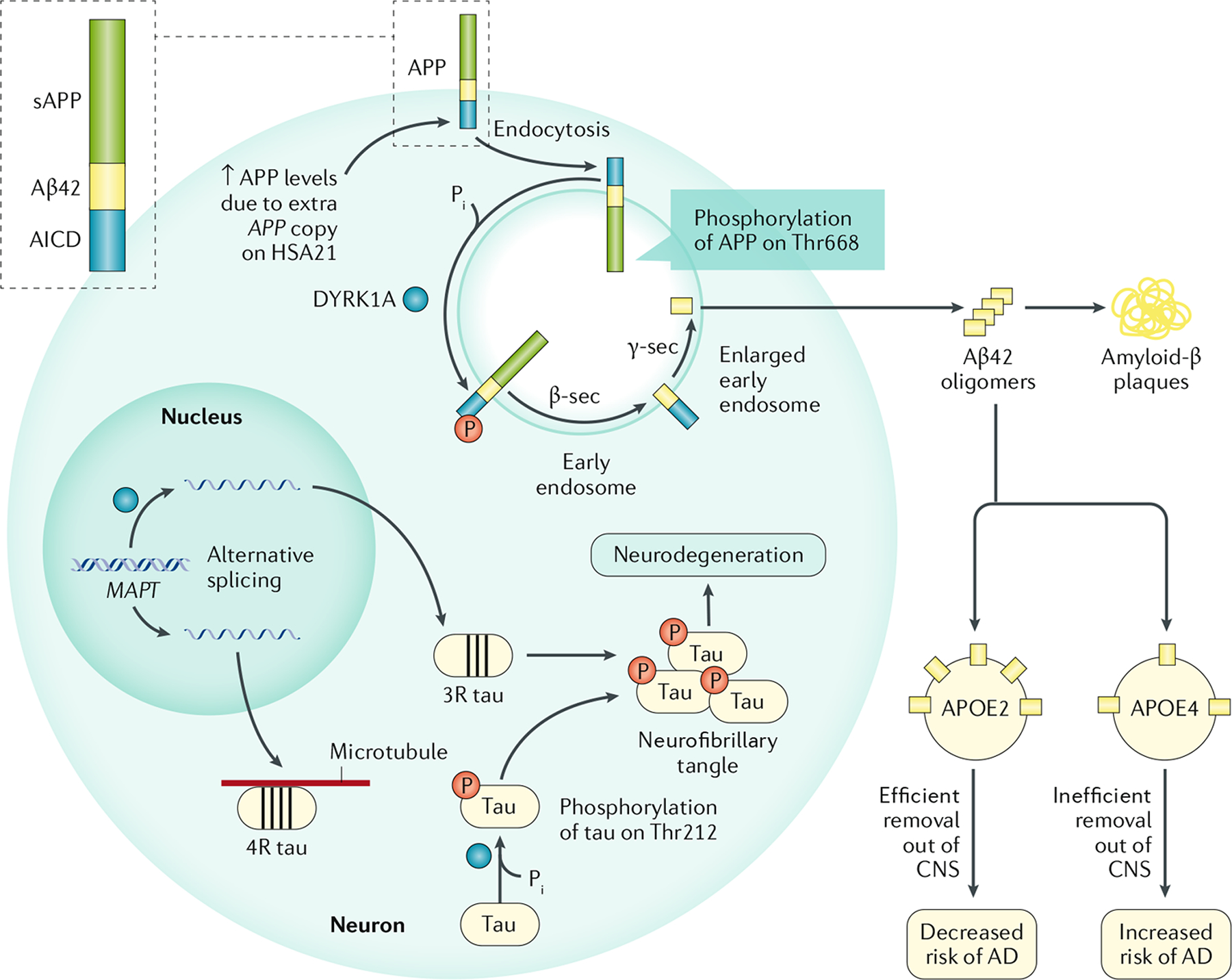
Trisomy 21, the presence of a supernumerary chromosome 21, results in a collection of clinical features commonly known as Down syndrome (DS). DS is among the most genetically complex of the conditions that are compatible with human survival post-term, and the most frequent survivable autosomal aneuploidy. Mouse models of DS, involving trisomy of all or part of human chromosome 21 or orthologous mouse genomic regions, are providing valuable insights into the contribution of triplicated genes or groups of genes to the many clinical manifestations in DS. This endeavour is challenging, as there are >200 protein-coding genes on chromosome 21 and they can have direct and indirect effects on homeostasis in cells, tissues, organs and systems. Although this complexity poses formidable challenges to understanding the underlying molecular basis for each of the many clinical features of DS, it also provides opportunities for improving understanding of genetic mechanisms underlying the development and function of many cell types, tissues, organs and systems. Since the first description of trisomy 21, we have learned much about intellectual disability and genetic risk factors for congenital heart disease. The lower occurrence of solid tumours in individuals with DS supports the identification of chromosome 21 genes that protect against cancer when overexpressed. The universal occurrence of the histopathology of Alzheimer disease and the high prevalence of dementia in DS are providing insights into the pathology and treatment of Alzheimer disease. Clinical trials to ameliorate intellectual disability in DS signal a new era in which therapeutic interventions based on knowledge of the molecular pathophysiology of DS can now be explored; these efforts provide reasonable hope for the future.
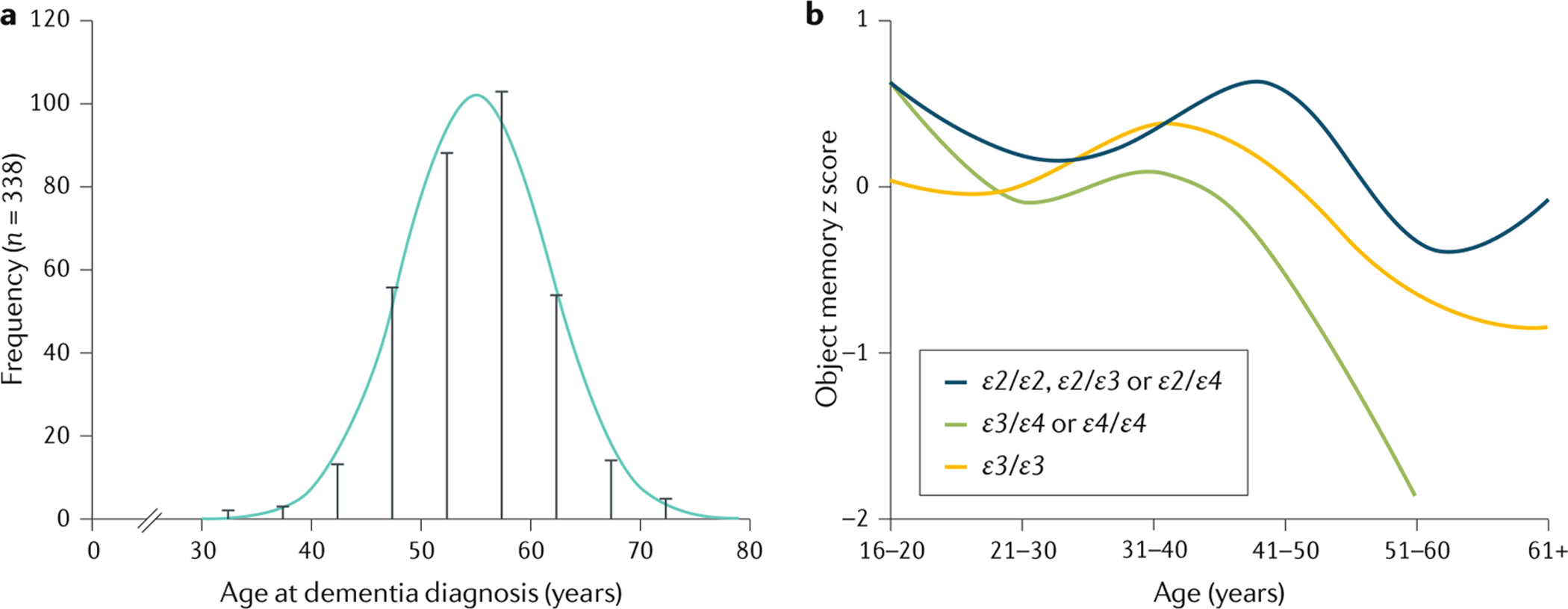
Figures
References
-
- Down JLH Observations on an ethnic classification of idiots. Lond. Hosp. Rep 3, 259–262 (1866).
-
- Hasle H, Clemmensen IH & Mikkelsen M Risks of leukaemia and solid tumours in individuals with Down’s syndrome. Lancet 355, 165–169 (2000). - PubMed
-
- LeJeune J, Gautier M & Turpin R Study of somatic chromosomes from 9 mongoloid children [French]. C. R. Hebd. Seances. Acad. Sci 248, 1721–1722 (1959). - PubMed
-
This study is the first description of the chromosomal abnormality in DS.
-
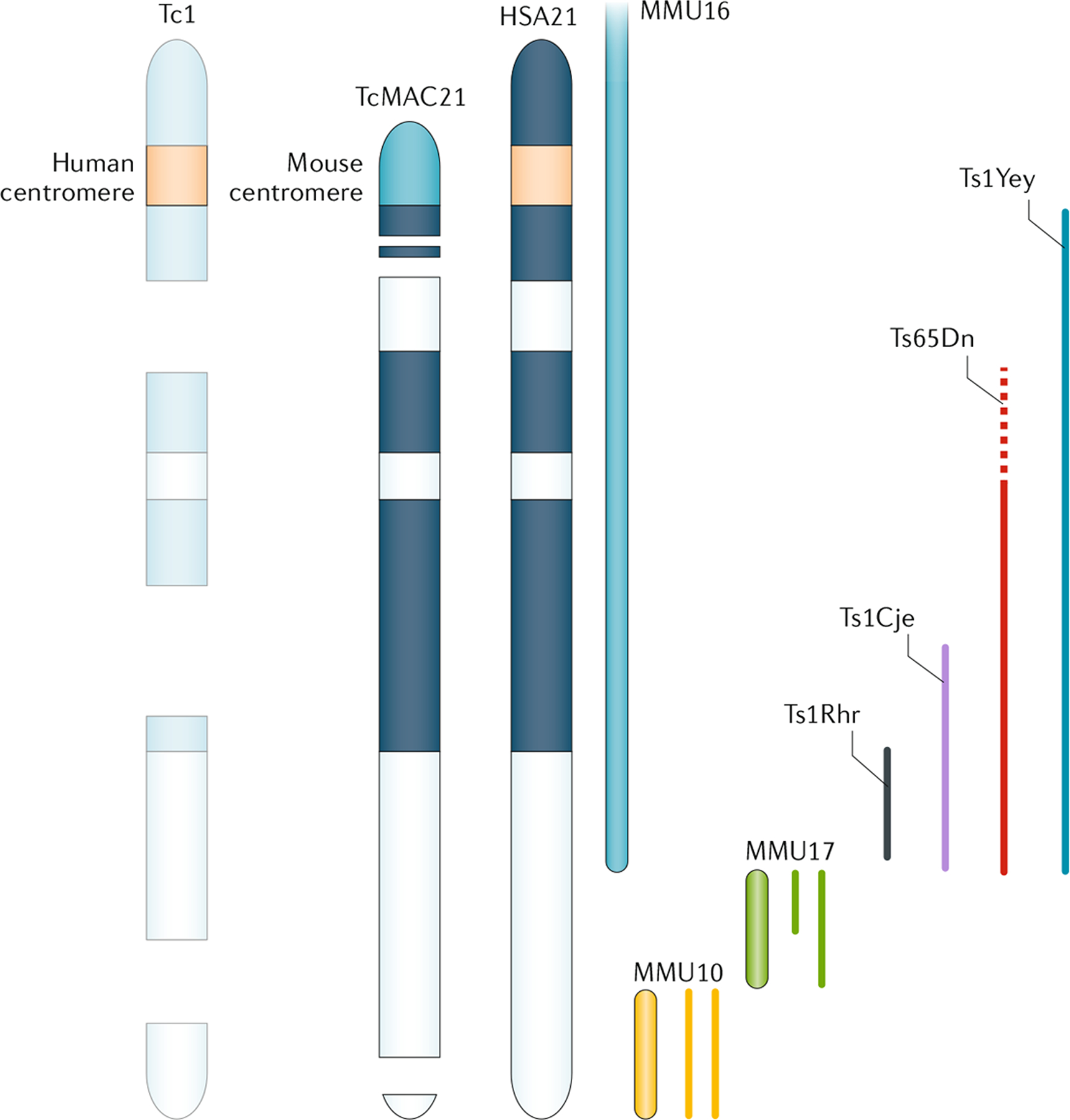
- Davisson MT, Schmidt C & Akeson EC Segmental trisomy of murine chromosome 16: a new model system for studying Down syndrome. Prog. Clin. Biol. Res 360, 263–280 (1990). - PubMed
-
- Hattori M et al. The DNA sequence of human chromosome 21. Nature 405, 311–319 (2000). - PubMed
-
A landmark paper on the sequencing of the long arm of HSA21.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
