

SmartPhase: Accurate and fast phasing of heterozygous variant pairs for genetic diagnosis of rare diseases
- PMID: 32032351
- PMCID: PMC7032729
- DOI: 10.1371/journal.pcbi.1007613
SmartPhase: Accurate and fast phasing of heterozygous variant pairs for genetic diagnosis of rare diseases
Abstract
There is an increasing need to use genome and transcriptome sequencing to genetically diagnose patients suffering from suspected monogenic rare diseases. The proper detection of compound heterozygous variant combinations as disease-causing candidates is a challenge in diagnostic workflows as haplotype information is lost by currently used next-generation sequencing technologies. Consequently, computational tools are required to phase, or resolve the haplotype of, the high number of heterozygous variants in the exome or genome of each patient. Here we present SmartPhase, a phasing tool designed to efficiently reduce the set of potential compound heterozygous variant pairs in genetic diagnoses pipelines. The phasing algorithm of SmartPhase creates haplotypes using both parental genotype information and reads generated by DNA or RNA sequencing and is thus well suited to resolve the phase of rare variants. To inform the user about the reliability of a phasing prediction, it computes a confidence score which is essential to select error-free predictions. It incorporates existing haplotype information and applies logical rules to determine variants that can be excluded as causing a recessive, monogenic disease. SmartPhase can phase either all possible variant pairs in predefined genetic loci or preselected variant pairs of interest, thus keeping the focus on clinically relevant results. We compared SmartPhase to WhatsHap, one of the leading comparable phasing tools, using simulated data and a real clinical cohort of 921 patients. On both data sets, SmartPhase generated error-free predictions using our derived confidence score threshold. It outperformed WhatsHap with regard to the percentage of resolved pairs when parental genotype information is available. On the cohort data, SmartPhase enabled on average the exclusion of approximately 22% of the input variant pairs in each singleton patient and 44% in each trio patient. SmartPhase is implemented as an open-source Java tool and freely available at http://ibis.helmholtz-muenchen.de/smartphase/.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
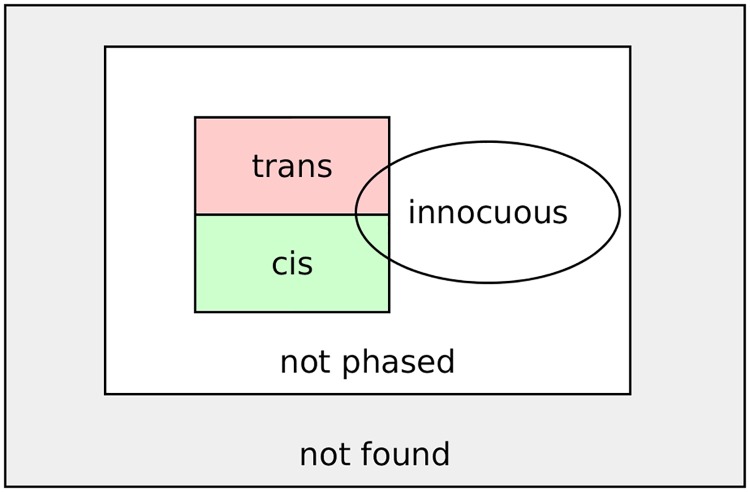
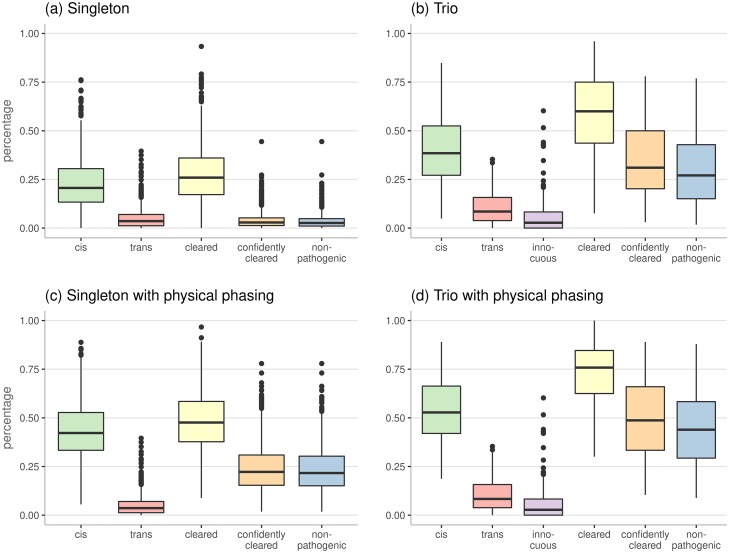
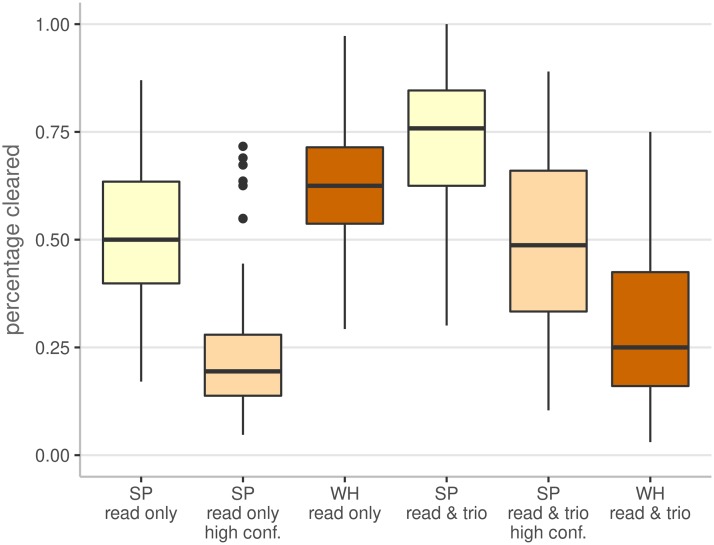
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
