

Modifier genes in SCN1A-related epilepsy syndromes
- PMID: 32032478
- PMCID: PMC7196470
- DOI: 10.1002/mgg3.1103
Modifier genes in SCN1A-related epilepsy syndromes
Abstract
Background: SCN1A is one of the most important epilepsy-related genes, with pathogenic variants leading to a range of phenotypes with varying disease severity. Different modifying factors have been hypothesized to influence SCN1A-related phenotypes. We investigate the presence of rare and more common variants in epilepsy-related genes as potential modifiers of SCN1A-related disease severity.
Methods: 87 patients with SCN1A-related epilepsy were investigated. Whole-exome sequencing was performed by the Beijing Genomics Institute (BGI). Functional variants in 422 genes associated with epilepsy and/or neuronal excitability were investigated. Differences in proportions of variants between the epilepsy genes and four control gene sets were calculated, and compared to the proportions of variants in the same genes in the ExAC database.
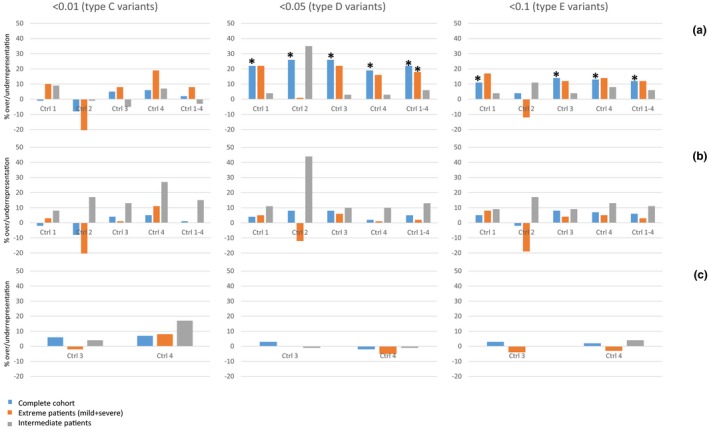
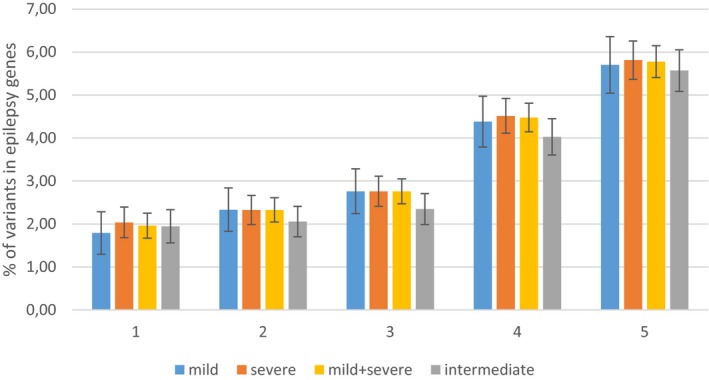
Results: Statistically significant excesses of variants in epilepsy genes were observed in the complete cohort and in the combined group of mildly and severely affected patients, particularly for variants with minor allele frequencies of <0.05. Patients with extreme phenotypes showed much greater excesses of epilepsy gene variants than patients with intermediate phenotypes.
Conclusion: Our results indicate that relatively common variants in epilepsy genes, which would not necessarily be classified as pathogenic, may play a large role in modulating SCN1A phenotypes. They may modify the phenotypes of both severely and mildly affected patients. Our results may be a first step toward meaningful testing of modifier gene variants in regular diagnostics for individual patients, to provide a better estimation of disease severity for newly diagnosed patients.
Keywords: SCN1A; Dravet; GEFS+; epilepsy; modifier genes; phenotypic variability.
© 2020 The Authors. Molecular Genetics & Genomic Medicine published by Wiley Periodicals, Inc.
Conflict of interest statement
None declared.
Figures
References
-
- Barrett, J. , Buxbaum, J. , Cutle, D. , Daly, M. , Devlin, B. , Gratten, J. , … Wray, N. (2017). New mutations, old statistical challenges Based. BioRxiv.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
