

The Canadian Rare Diseases Models and Mechanisms (RDMM) Network: Connecting Understudied Genes to Model Organisms
- PMID: 32032513
- PMCID: PMC7010971
- DOI: 10.1016/j.ajhg.2020.01.009
The Canadian Rare Diseases Models and Mechanisms (RDMM) Network: Connecting Understudied Genes to Model Organisms
Abstract
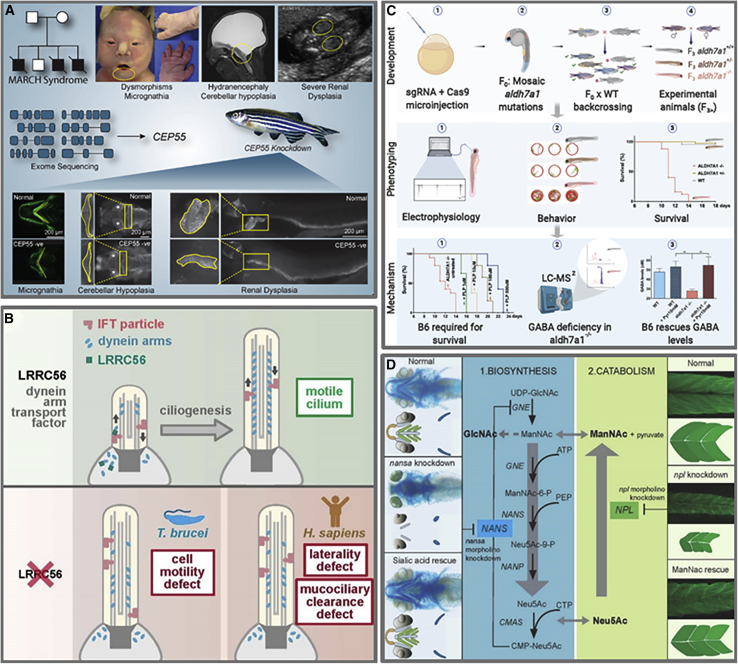
Advances in genomics have transformed our ability to identify the genetic causes of rare diseases (RDs), yet we have a limited understanding of the mechanistic roles of most genes in health and disease. When a novel RD gene is first discovered, there is minimal insight into its biological function, the pathogenic mechanisms of disease-causing variants, and how therapy might be approached. To address this gap, the Canadian Rare Diseases Models and Mechanisms (RDMM) Network was established to connect clinicians discovering new disease genes with Canadian scientists able to study equivalent genes and pathways in model organisms (MOs). The Network is built around a registry of more than 500 Canadian MO scientists, representing expertise for over 7,500 human genes. RDMM uses a committee process to identify and evaluate clinician-MO scientist collaborations and approve 25,000 Canadian dollars in catalyst funding. To date, we have made 85 clinician-MO scientist connections and funded 105 projects. These collaborations help confirm variant pathogenicity and unravel the molecular mechanisms of RD, and also test novel therapies and lead to long-term collaborations. To expand the impact and reach of this model, we made the RDMM Registry open-source, portable, and customizable, and we freely share our committee structures and processes. We are currently working with emerging networks in Europe, Australia, and Japan to link international RDMM networks and registries and enable matches across borders. We will continue to create meaningful collaborations, generate knowledge, and advance RD research locally and globally for the benefit of patients and families living with RD.
Keywords: functional insight; gene discovery; model organisms; rare genetic diseases.
Copyright © 2020 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declarations of Interest The authors declare no competing interests.
Figures



References
-
- Hartley T., Balcı T.B., Rojas S.K., Eaton A., Canada C.R., Dyment D.A., Boycott K.M. The unsolved rare genetic disease atlas? An analysis of the unexplained phenotypic descriptions in OMIM®. Am. J. Med. Genet. C. Semin. Med. Genet. 2018;178:458–463. - PubMed
-
- Dolinski K., Botstein D. Orthology and functional conservation in eukaryotes. Annu. Rev. Genet. 2007;41:465–507. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
