

The green tea polyphenol epigallocatechin-3-gallate (EGCG) restores CDKL5-dependent synaptic defects in vitro and in vivo
- PMID: 32032735
- PMCID: PMC7152796
- DOI: 10.1016/j.nbd.2020.104791
The green tea polyphenol epigallocatechin-3-gallate (EGCG) restores CDKL5-dependent synaptic defects in vitro and in vivo
Abstract
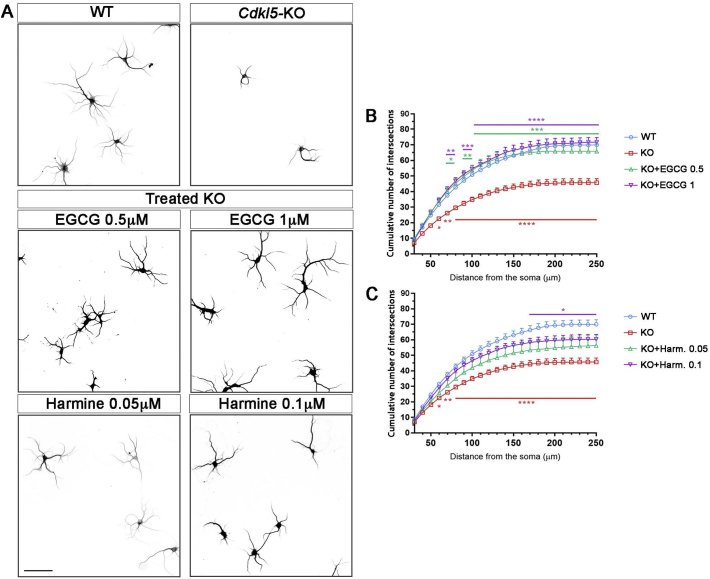
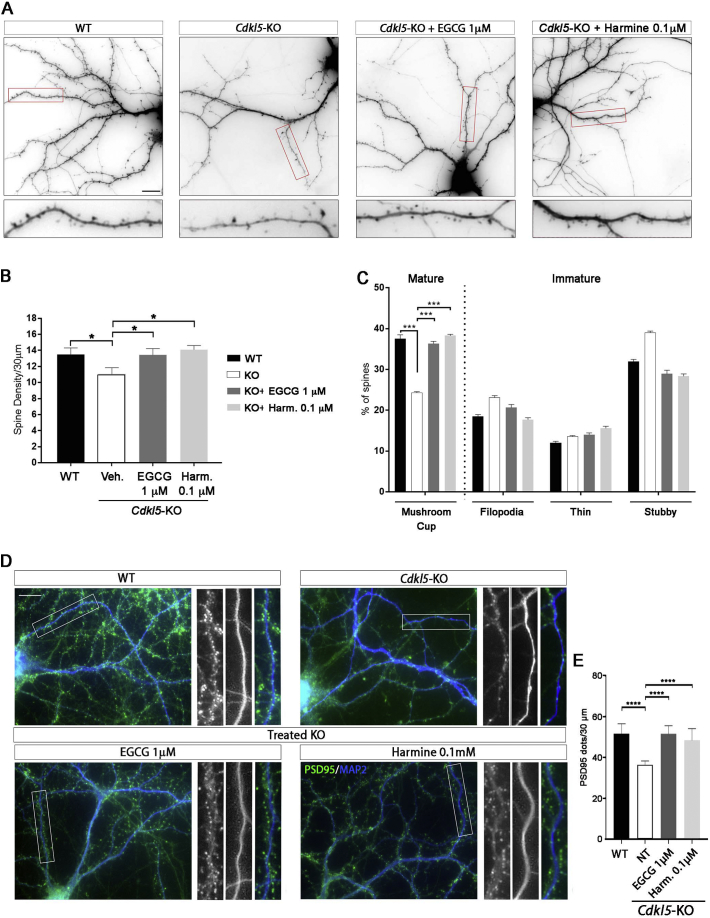
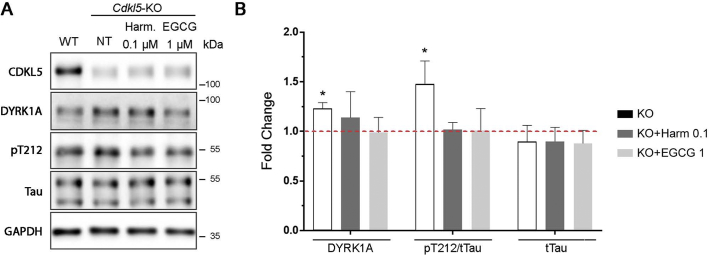
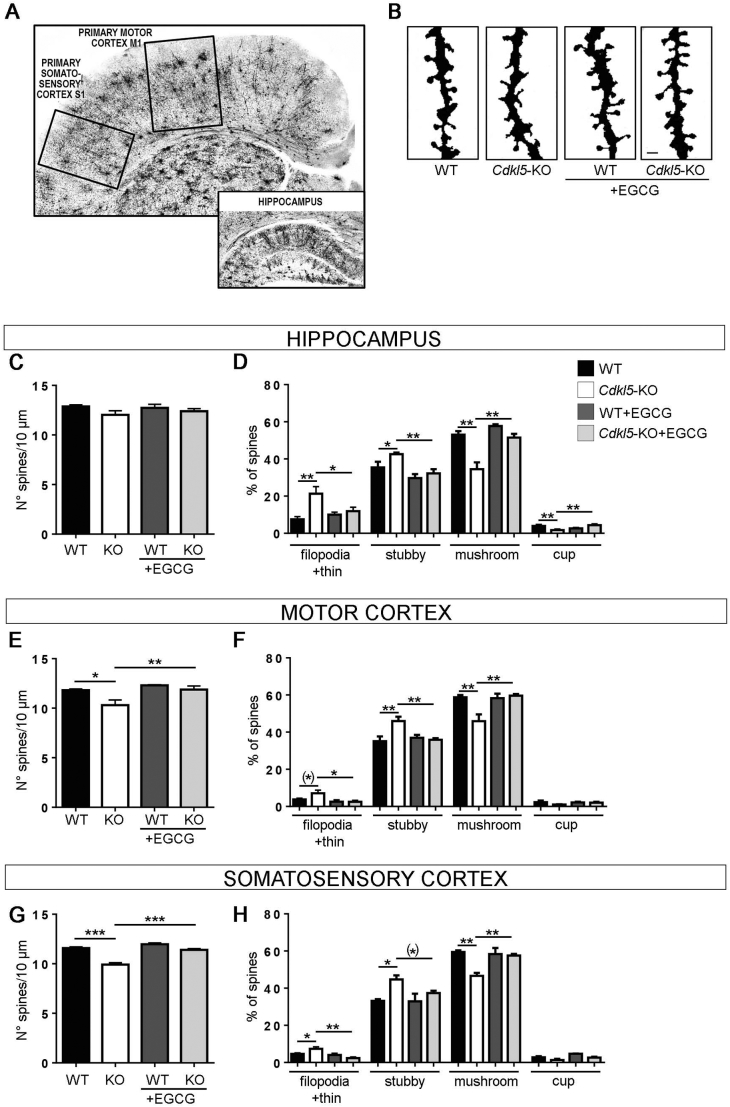
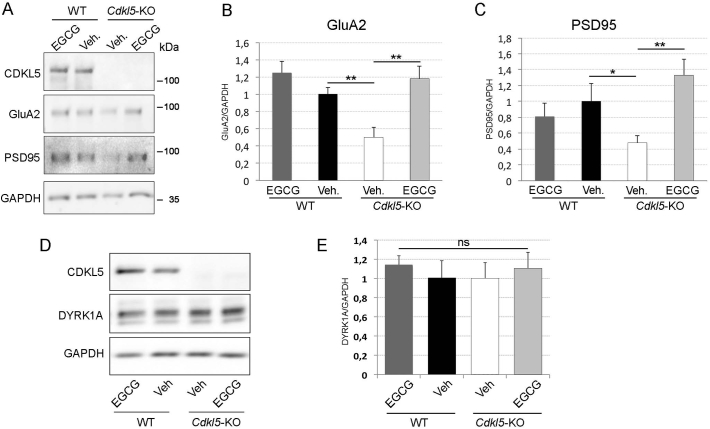
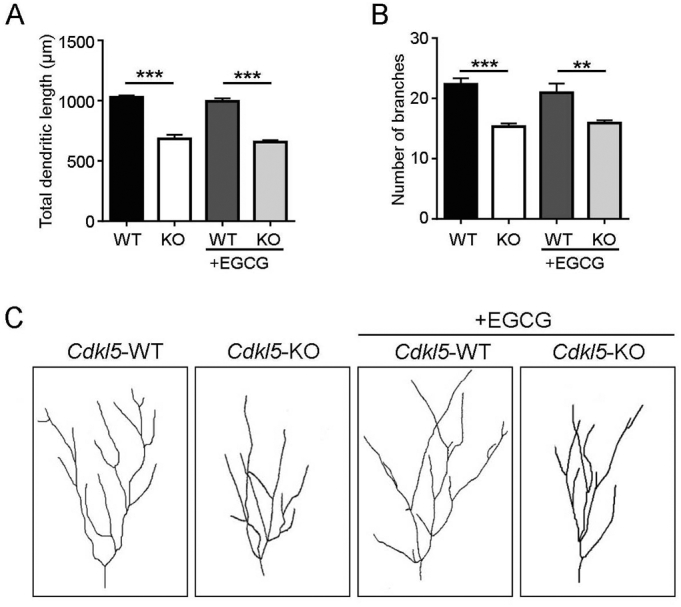
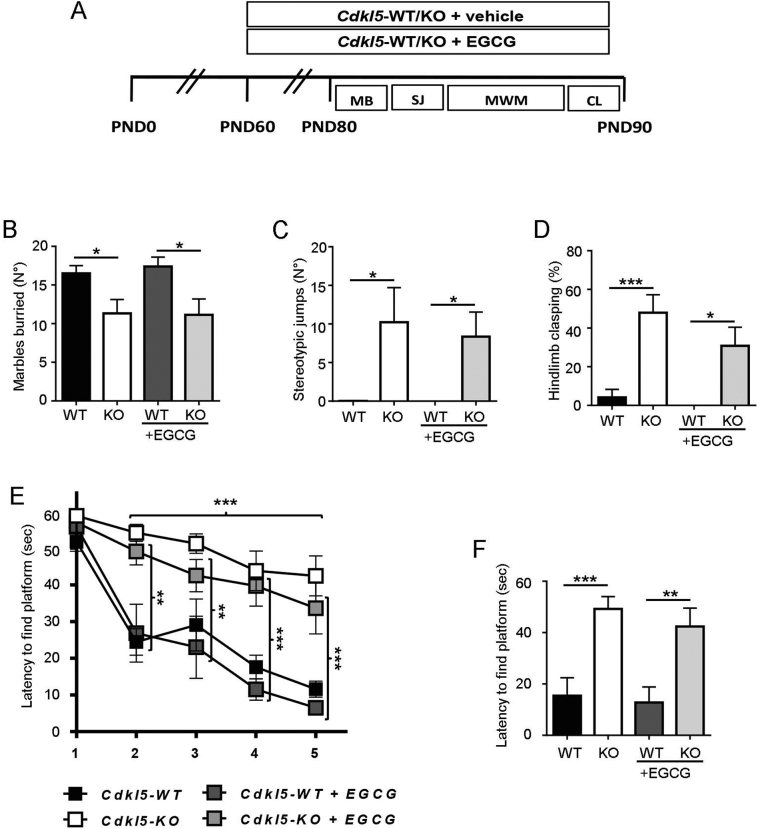
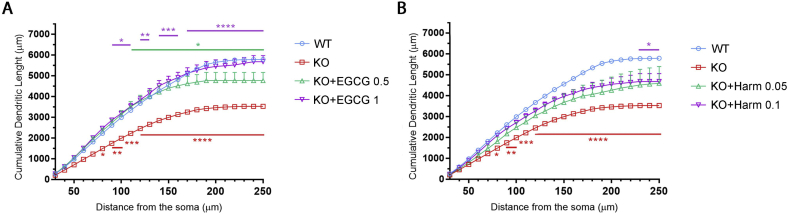
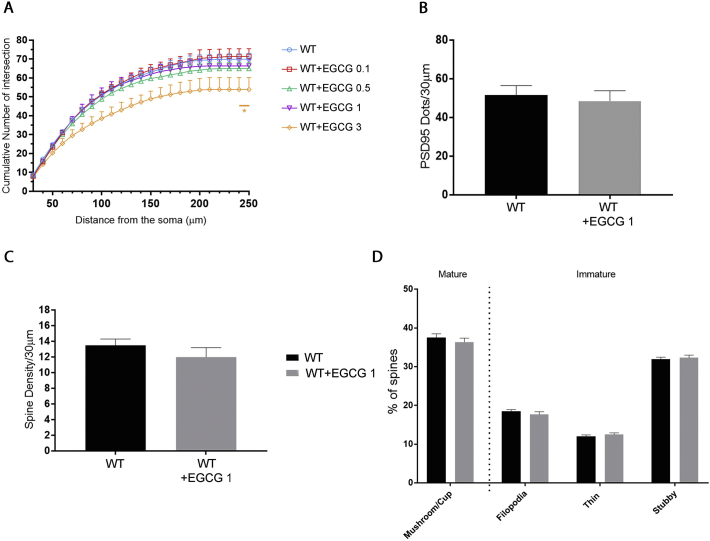
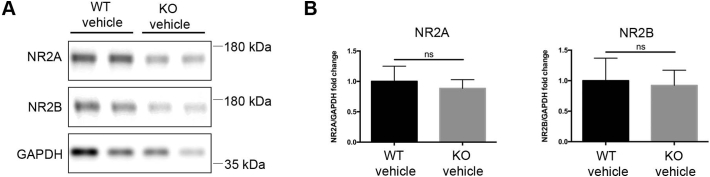
CDKL5 deficiency disorder (CDD) is a rare X-linked neurodevelopmental disorder that is characterised by early-onset seizures, intellectual disability, gross motor impairment, and autistic-like features. CDD is caused by mutations in the cyclin-dependent kinase-like 5 (CDKL5) gene that encodes a serine/threonine kinase with a predominant expression in the brain. Loss of CDKL5 causes neurodevelopmental alterations in vitro and in vivo, including defective dendritic arborisation and spine maturation, which most likely underlie the cognitive defects and autistic features present in humans and mice. Here, we show that treatment with epigallatocathechin-3-gallate (EGCG), the major polyphenol of green tea, can restore defects in dendritic and synaptic development of primary Cdkl5 knockout (KO) neurons. Furthermore, defective synaptic maturation in the hippocampi and cortices of adult Cdkl5-KO mice can be rescued through the intraperitoneal administration of EGCG, which is however not sufficient to normalise behavioural CDKL5-dependent deficits. EGCG is a pleiotropic compound with numerous cellular targets, including the dual-specificity tyrosine-phosphorylation-regulated kinase 1A (DYRK1A) that is selectively inhibited by EGCG. DYRK1A controls dendritic development and spine formation and its deregulation has been implicated in neurodevelopmental and degenerative diseases. Treatment with another DYRK1A inhibitor, harmine, was capable of correcting neuronal CDKL5-dependent defects; moreover, DYRK1A levels were upregulated in primary Cdkl5-KO neurons in concomitance with increased phosphorylation of Tau, a well-accepted DYRK1A substrate. Altogether, our results indicate that DYRK1A deregulation may contribute, at least in part, to the neurodevelopmental alterations caused by CDKL5 deficiency.
Keywords: CDKL5; DYRK1A; Epigallatocathechin-3-gallate; Synaptic defects.
Copyright © 2020 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Competing Interest None. The following are the supplementary data related to this article. Supplementary data to this article can be found online at https://doi.org/10.1016/j.nbd.2020.104791.
Figures
References
-
- Arbones M.L., Thomazeau A., Nakano-Kobayashi A., Hagiwara M., Delabar J.M. DYRK1A and cognition: a lifelong relationship. Pharmacol. Ther. 2019 - PubMed
-
- Bertani I., Rusconi L., Bolognese F., Forlani G., Conca B., De Monte L., Badaracco G., Landsberger N., Kilstrup-Nielsen C. Functional consequences of mutations in CDKL5, an X-linked gene involved in infantile spasms and mental retardation. J. Biol. Chem. 2006;281:32048–32056. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
