

Chronic Oral Palmitoylethanolamide Administration Rescues Cognitive Deficit and Reduces Neuroinflammation, Oxidative Stress, and Glutamate Levels in A Transgenic Murine Model of Alzheimer's Disease
- PMID: 32033363
- PMCID: PMC7074257
- DOI: 10.3390/jcm9020428
Chronic Oral Palmitoylethanolamide Administration Rescues Cognitive Deficit and Reduces Neuroinflammation, Oxidative Stress, and Glutamate Levels in A Transgenic Murine Model of Alzheimer's Disease
Abstract
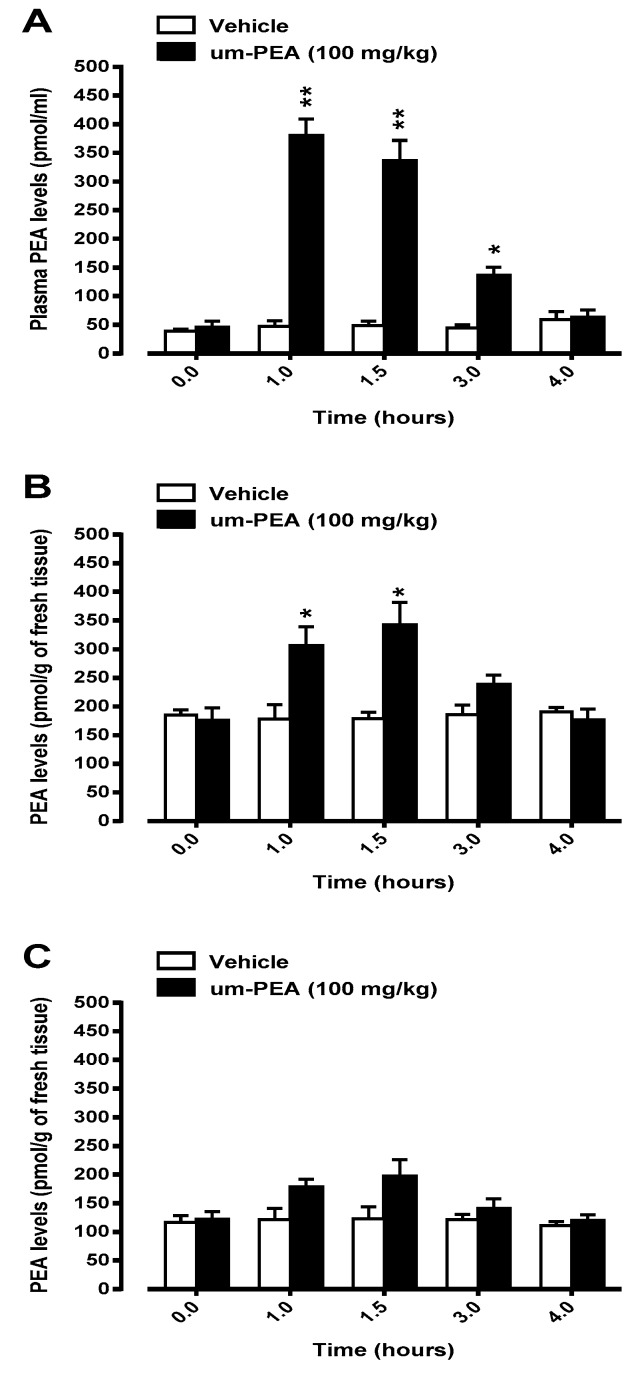
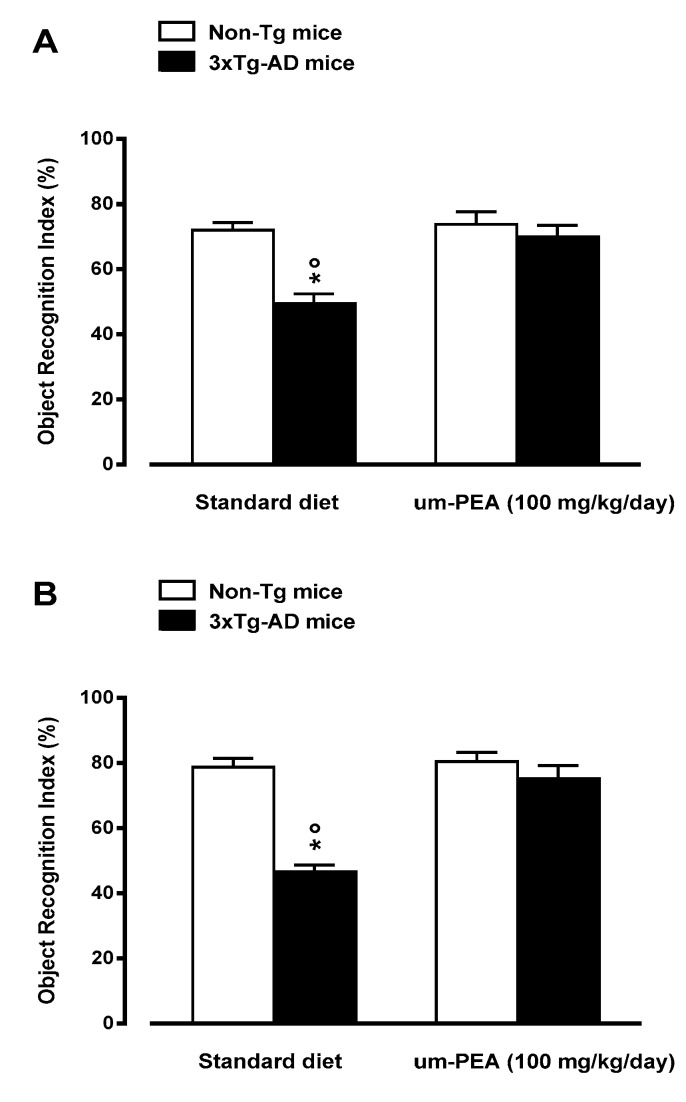
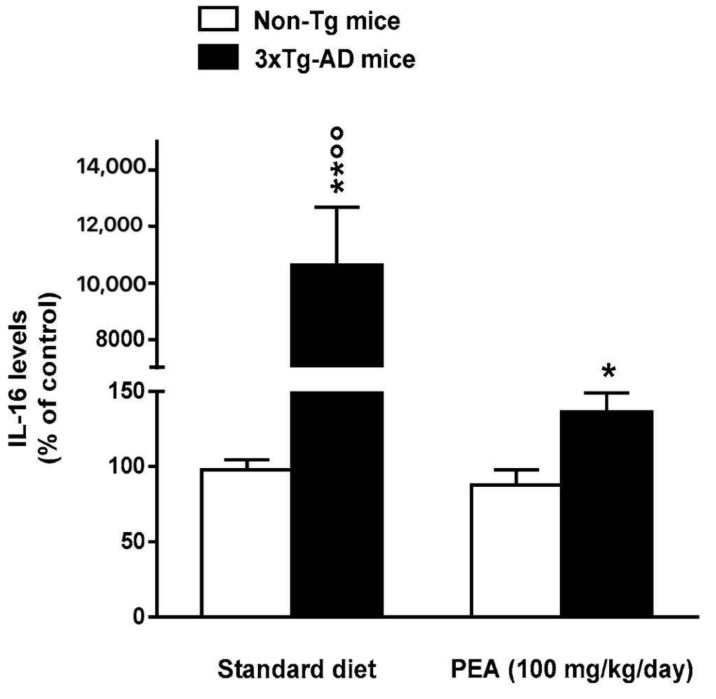
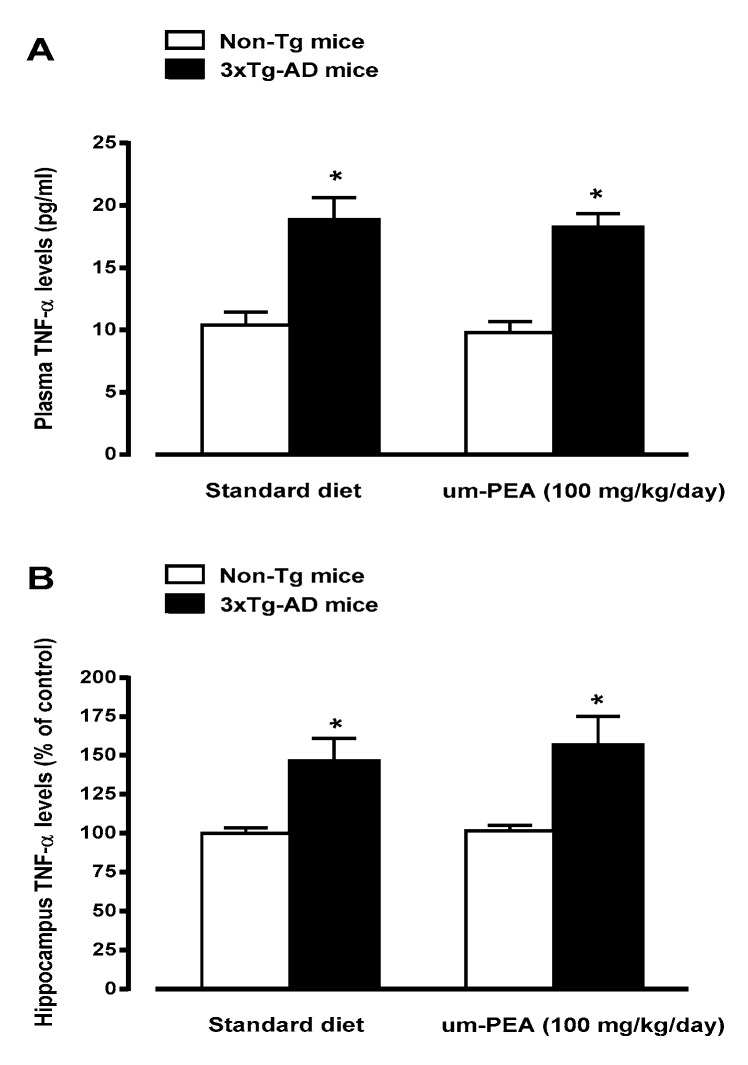
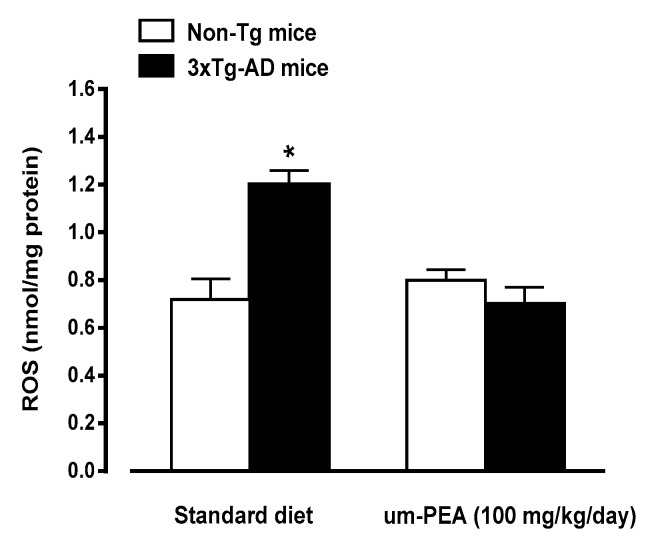
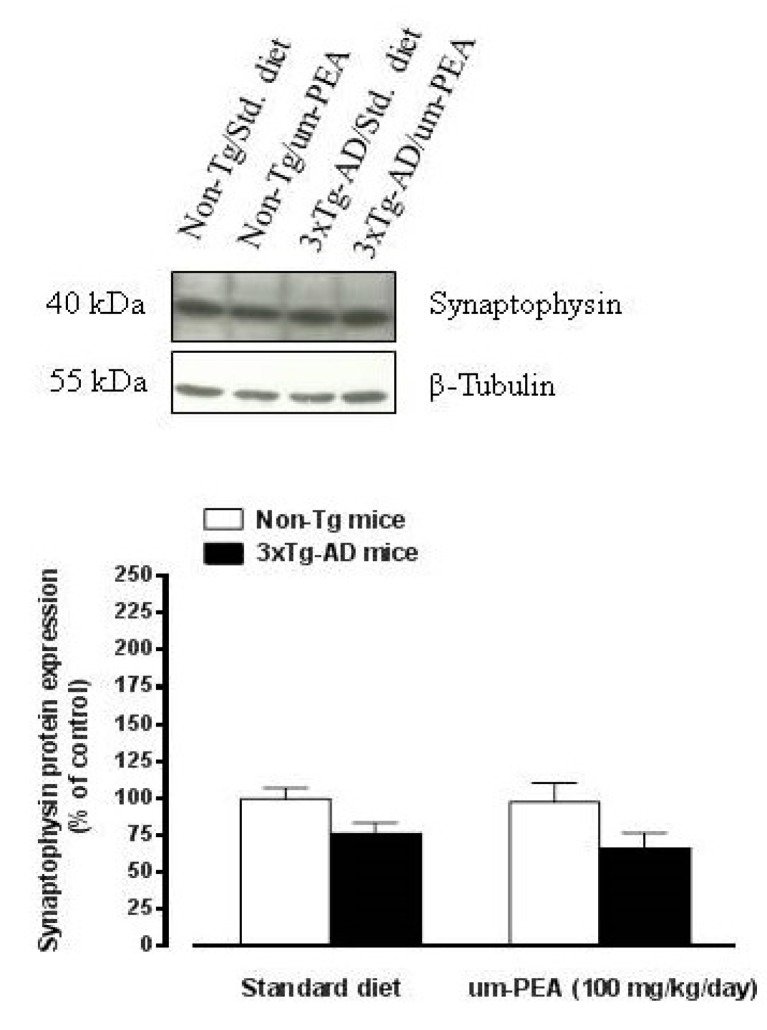
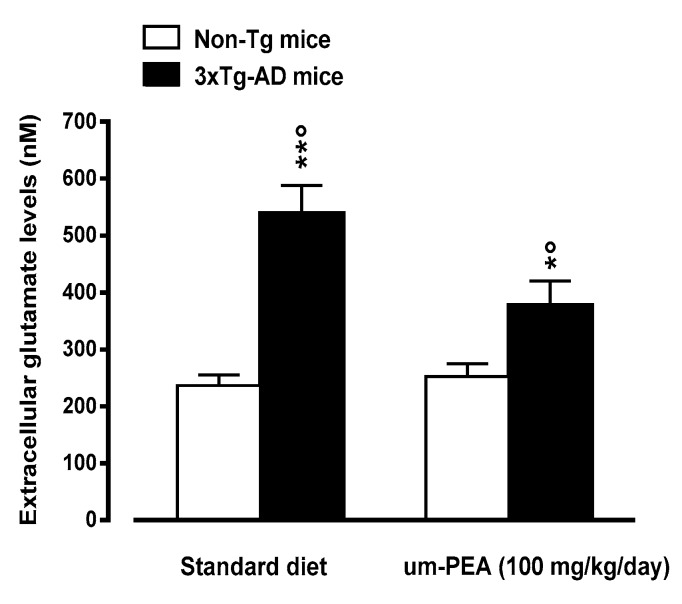
N-palmitoylethanolamide (PEA) is a lipid mediator belonging to the class of the N-acylethanolamine. Products containing PEA, also in ultramicronized formulation (um-PEA), are already licensed for use in humans for its analgesic and anti-inflammatory properties, and demonstrated high safety and tolerability. Preclinical studies indicate that PEA, especially in the ultramicronized form, could be a potential therapeutic agent for Alzheimer's disease (AD). In this study, we evaluated the neuroprotective and antioxidant effects of chronic (three months) um-PEA administration in an animal model of AD (3×Tg-AD mice). For translation purposes, the compound has been orally administered. Cognitive performance as well as biochemical markers [(interleukin-16 (IL-16) and tumor necrosis factor- (TNF-)] levels, reactive oxygen species (ROS) production, synaptophysin and glutamate levels) have been evaluated at the end of um-PEA treatment. The results indicate that orally administered um-PEA was adsorbed and distributed in the mice brain. The chronic treatment with um-PEA (100 mg/kg/day for three months) rescued cognitive deficit, restrained neuroinflammation and oxidative stress, and reduced the increase in hippocampal glutamate levels observed in 3×Tg-AD mice. Overall, these data reinforce the concept that um-PEA exerts beneficial effects in 3×Tg-AD mice. The fact that PEA is already licensed for the use in humans strongly supports its rapid translation in clinical practice.
Keywords: Alzheimer’s disease; cognitive dysfunctions; extracellular glutamate levels; hippocampus; reactive oxygen species; synaptophysin.
Conflict of interest statement
No conflict of interest.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
