

Eleven grand challenges in single-cell data science
- PMID: 32033589
- PMCID: PMC7007675
- DOI: 10.1186/s13059-020-1926-6
Eleven grand challenges in single-cell data science
Abstract
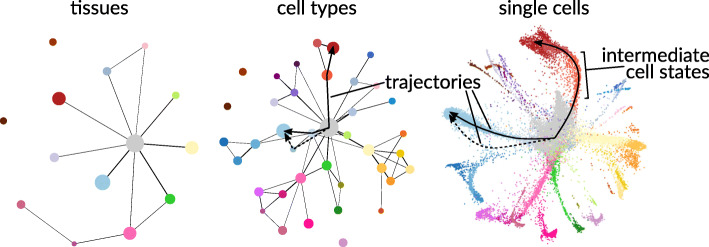
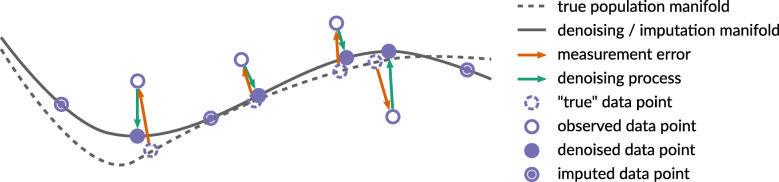
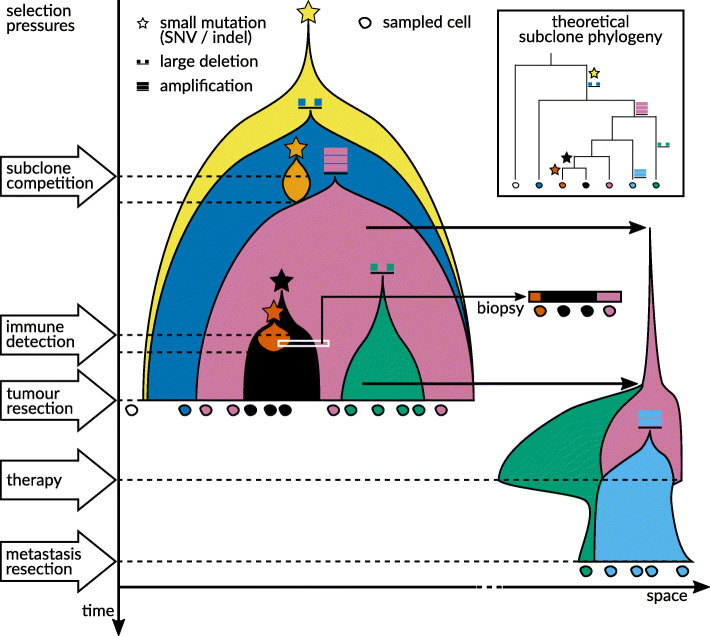
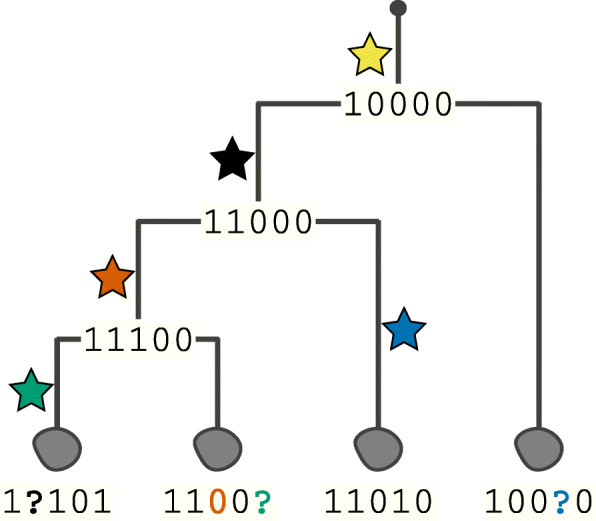
The recent boom in microfluidics and combinatorial indexing strategies, combined with low sequencing costs, has empowered single-cell sequencing technology. Thousands-or even millions-of cells analyzed in a single experiment amount to a data revolution in single-cell biology and pose unique data science problems. Here, we outline eleven challenges that will be central to bringing this emerging field of single-cell data science forward. For each challenge, we highlight motivating research questions, review prior work, and formulate open problems. This compendium is for established researchers, newcomers, and students alike, highlighting interesting and rewarding problems for the coming years.
Conflict of interest statement
BJR is a co-founder and consultant at Medley Genomics. FJT reports receiving consulting fees from Roche Diagnostics GmbH and Cellarity Inc., and ownership interest in Cellarity, Inc. and Dermagnostix GmbH. IIM is a co-founder and holds an interest in SmplBio LLC, a company developing cloud-based scRNA-Seq analysis software. No products, services, or technologies of SmplBio have been evaluated or tested in this work. JdR is co-founder of Cyclomics BV. SA is a scientific advisor to Sangamo and Repare Therapeutics. SA and SPS are both founders and shareholders of Contextual Genomics Inc. All other authors declare that they have no competing interests.
Figures

References
-
- Francis JM, Zhang C-Z, Maire CL, Jung J, Manzo VE, Adalsteinsson VA, Homer H, Haidar S, Blumenstiel B, Pedamallu CS, Ligon AH, Love JC, Meyerson M, Ligon KL. EGFR variant heterogeneity in glioblastoma resolved through single-nucleus sequencing. Cancer Discov. 2014;4(8):956–71. doi: 10.1158/2159-8290.CD-13-0879. - DOI - PMC - PubMed
-
- Regev A, Teichmann SA, Lander ES, Amit I, Benoist C, Birney E, Bodenmiller B, Campbell P, Carninci P, Clatworthy M, Clevers H, Deplancke B, Dunham I, Eberwine J, Eils R, Enard W, Farmer A, Fugger L, Göttgens B, Hacohen N, Haniffa M, Hemberg M, Kim S, Klenerman P, Kriegstein A, Lein E, Linnarsson S, Lundeberg J, Majumder P, Marioni JC, Merad M, Mhlanga M, Nawijn M, Netea M, Nolan G, Pe’er D, Phillipakis A, Ponting CP, Quake S, Reik W, Rozenblatt-Rosen O, Sanes J, Satija R, Schumacher TN, Shalek A, Shapiro E, Sharma P, Shin JW, Stegle O, Stratton M, Stubbington MJT, Oudenaarden AV, Wagner A, Watt F, Weissman J, Wold B, Xavier R, Yosef N, et al.The Human Cell Atlas. 2017. 10.1101/121202. Accessed 27 Mar 2019.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
