

Exome sequencing revealed DNA variants in NCOR1, IGF2BP1, SGLT2 and NEK11 as potential novel causes of ketotic hypoglycemia in children
- PMID: 32034166
- PMCID: PMC7005888
- DOI: 10.1038/s41598-020-58845-3
Exome sequencing revealed DNA variants in NCOR1, IGF2BP1, SGLT2 and NEK11 as potential novel causes of ketotic hypoglycemia in children
Abstract
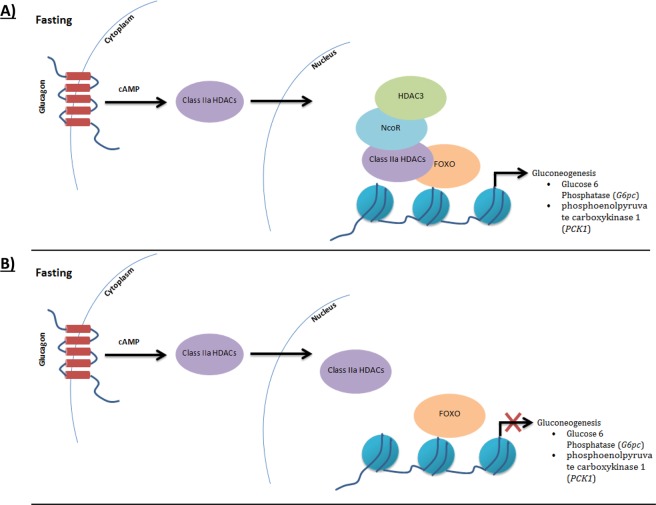
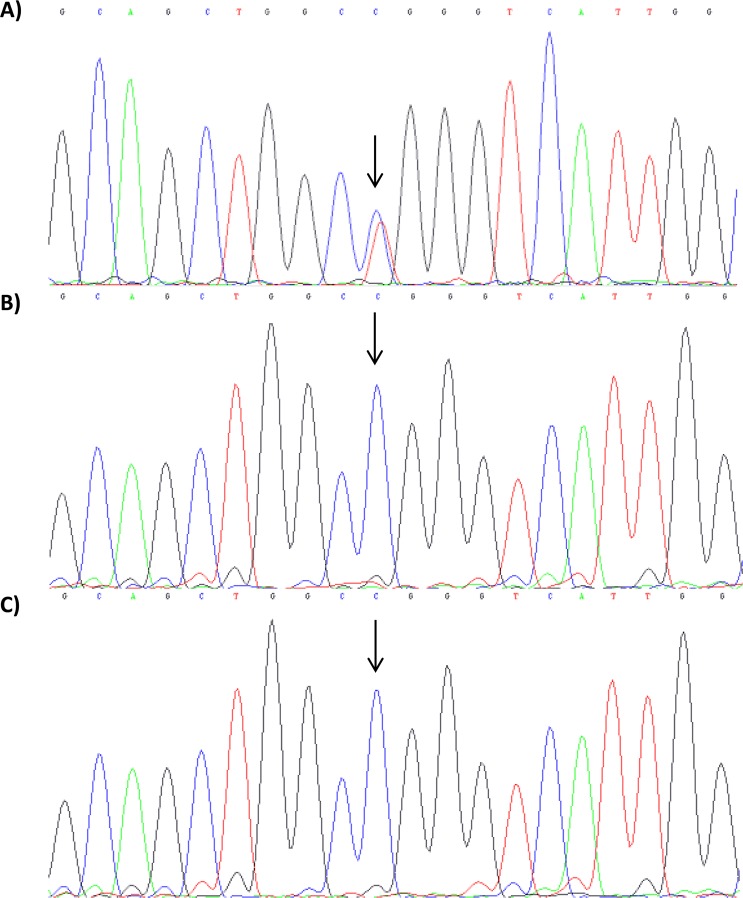
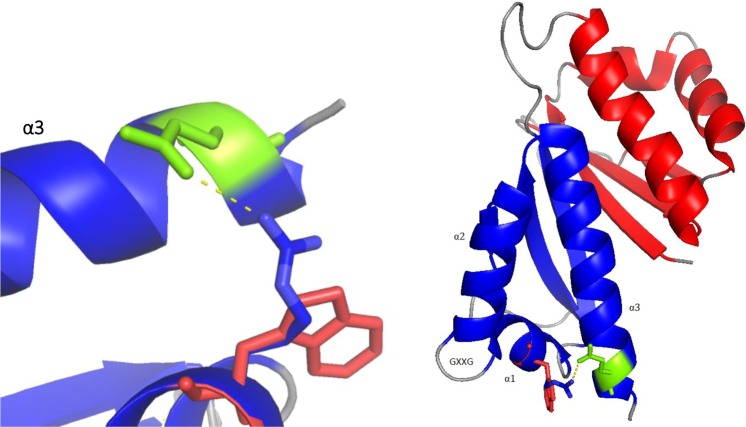
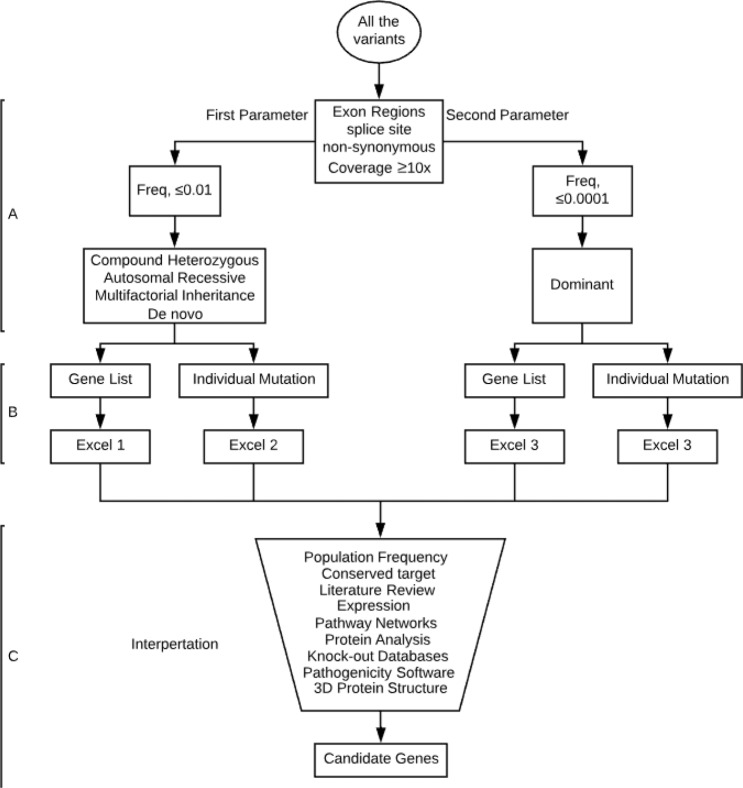
Unexplained or idiopathic ketotic hypoglycemia (KH) is the most common type of hypoglycemia in children. The diagnosis is based on the exclusion of routine hormonal and metabolic causes of hypoglycemia. We aimed to identify novel genes that cause KH, as this may lead to a more targeted treatment. Deep phenotyping of ten preschool age at onset KH patients (boys, n = 5; girls, n = 5) was performed followed by trio exome sequencing and comprehensive bioinformatics analysis. Data analysis revealed four novel candidate genes: (1) NCOR1 in a patient with KH, iron deficiency and loose stools; (2) IGF2BP1 in a proband with KH, short stature and delayed bone age; (3) SLC5A2 in a proband with KH, intermittent glucosuria and extremely elevated p-GLP-1; and (4) NEK11 in a proband with ketotic hypoglycemia and liver affliction. These genes are associated with different metabolic processes, such as gluconeogenesis, translational regulation, and glucose transport. In conclusion, WES identified DNA variants in four different genes as potential novel causes of IKH, suggesting that IKH is a heterogeneous disorder that can be split into several novel diseases: NCOR1-KH, IGF2BP1-KH, SGLT2-KH or familial renal glucosuria KH, and NEK11-KH. Precision medicine treatment based on exome sequencing may lead to advances in the management of IKH.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Andersen UM, Lund HT. [Hypoglycemia caused by growth hormone deficiency. Two cases in children with cerebral paresis]. Ugeskr. Laeger. 1995;157:1681–1682. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
