

Pan-genomics in the human genome era
- PMID: 32034321
- PMCID: PMC7752153
- DOI: 10.1038/s41576-020-0210-7
Pan-genomics in the human genome era
Abstract
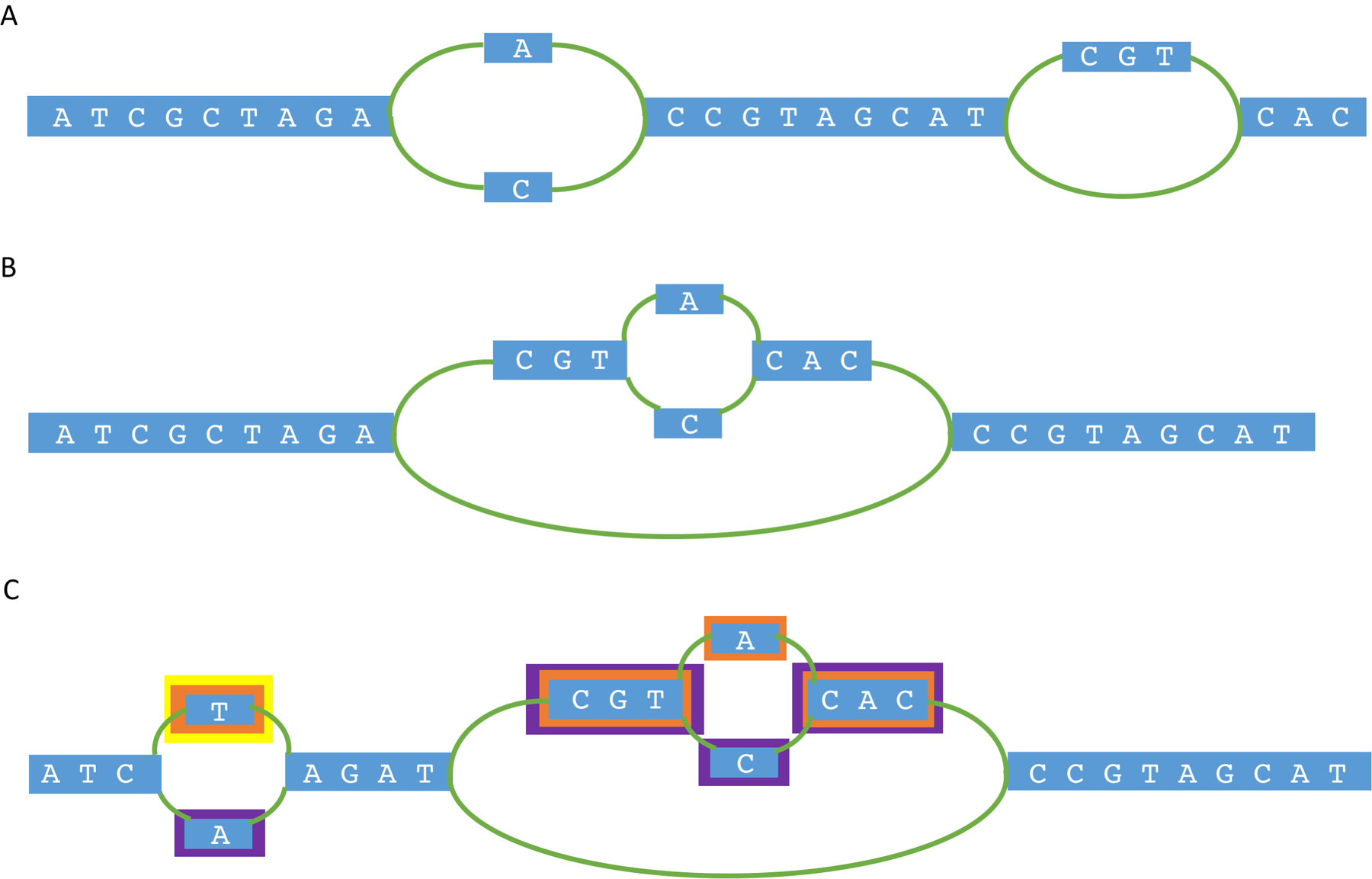
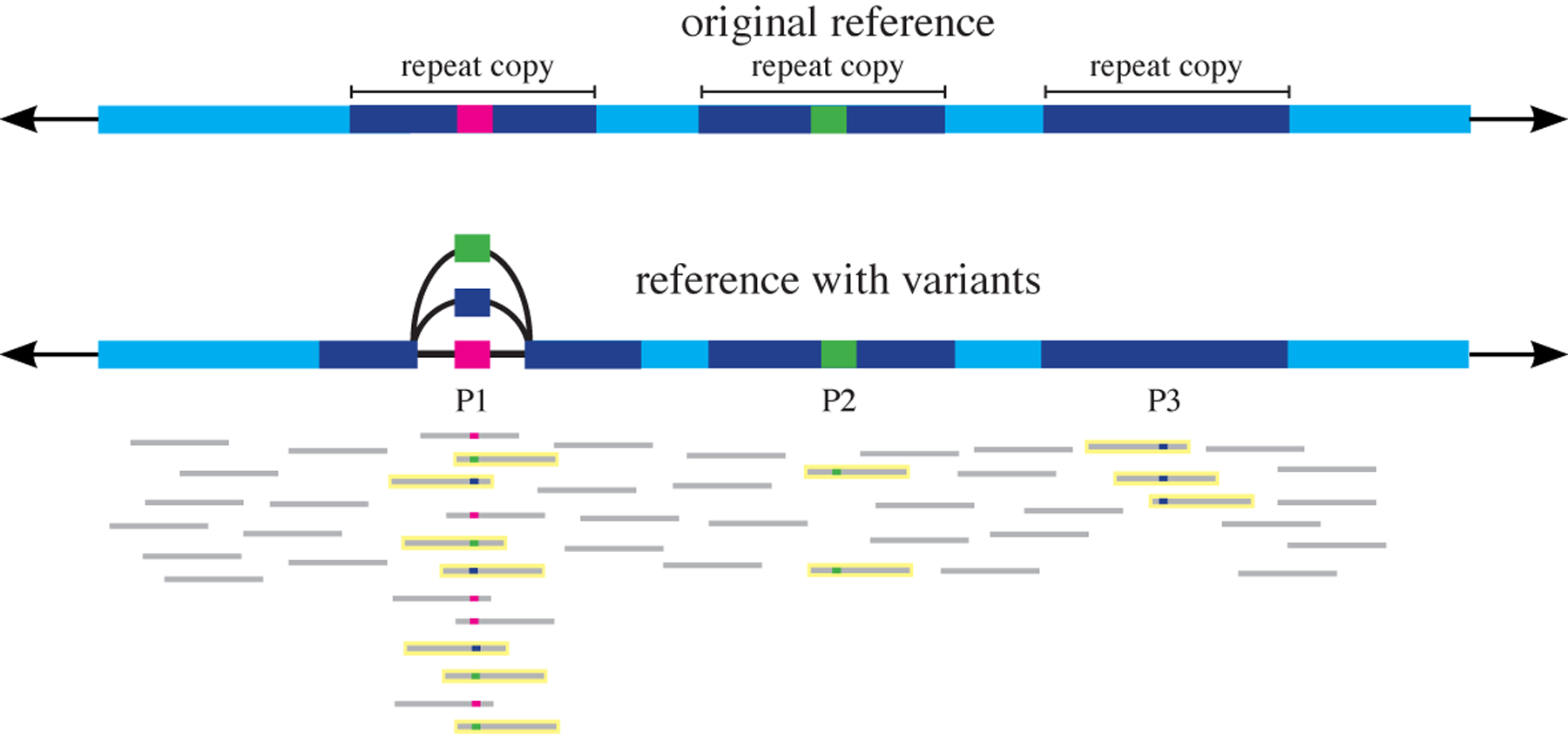
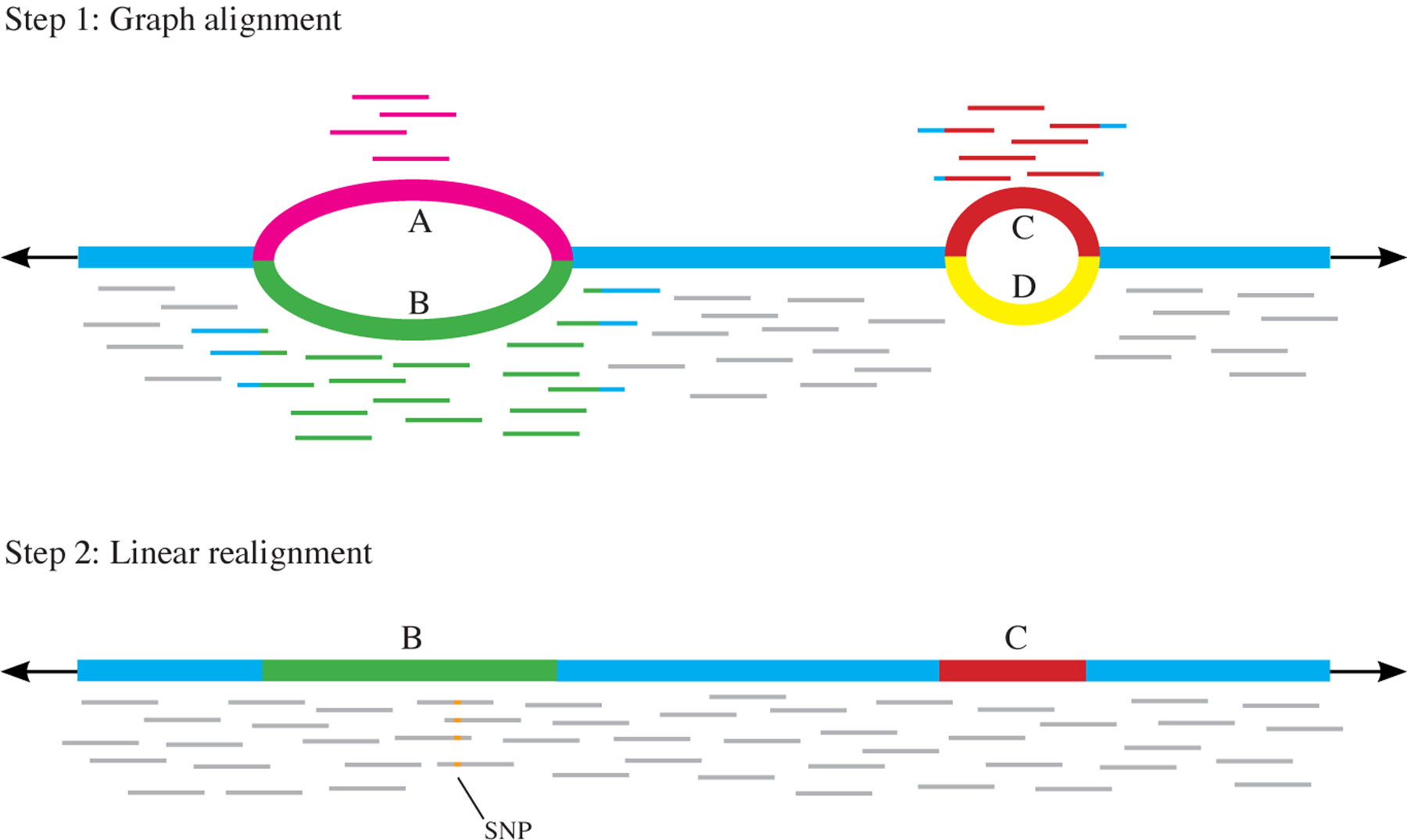
Since the early days of the genome era, the scientific community has relied on a single 'reference' genome for each species, which is used as the basis for a wide range of genetic analyses, including studies of variation within and across species. As sequencing costs have dropped, thousands of new genomes have been sequenced, and scientists have come to realize that a single reference genome is inadequate for many purposes. By sampling a diverse set of individuals, one can begin to assemble a pan-genome: a collection of all the DNA sequences that occur in a species. Here we review efforts to create pan-genomes for a range of species, from bacteria to humans, and we further consider the computational methods that have been proposed in order to capture, interpret and compare pan-genome data. As scientists continue to survey and catalogue the genomic variation across human populations and begin to assemble a human pan-genome, these efforts will increase our power to connect variation to human diversity, disease and beyond.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures
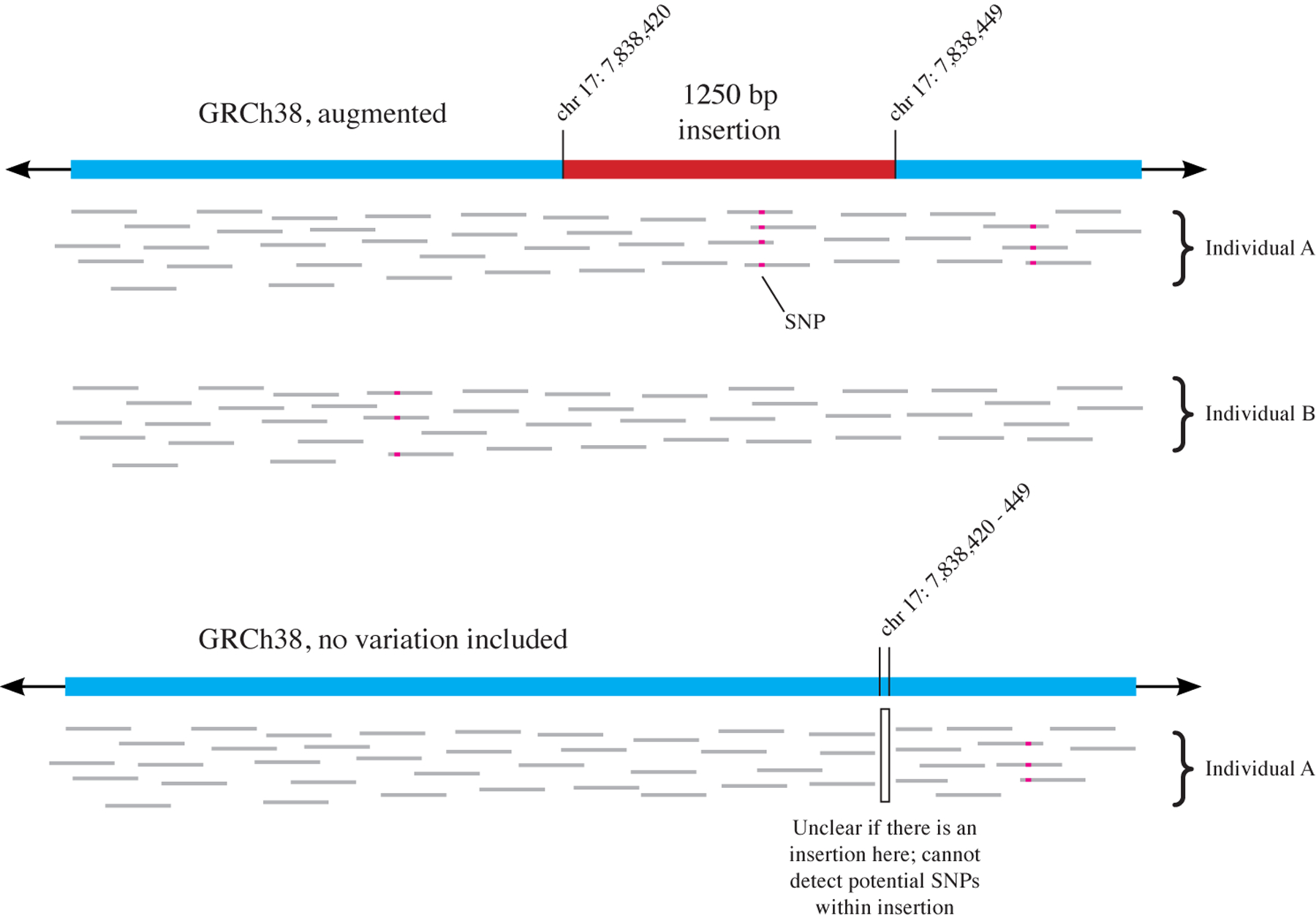
References
-
- National Human Genome Reserach Institute. Human Genome Project FAQ. NIH https://www.genome.gov/human-genome-project/Completion-FAQ (2019).
-
- Pallen MJ & Wren BW Bacterial pathogenomics. Nature 449, 835–842 (2007). - PubMed
-
- Tettelin H et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial ‘pan-genome’. Proc. Natl Acad. Sci. USA 102, 13950–13955 (2005). - PMC - PubMed
-
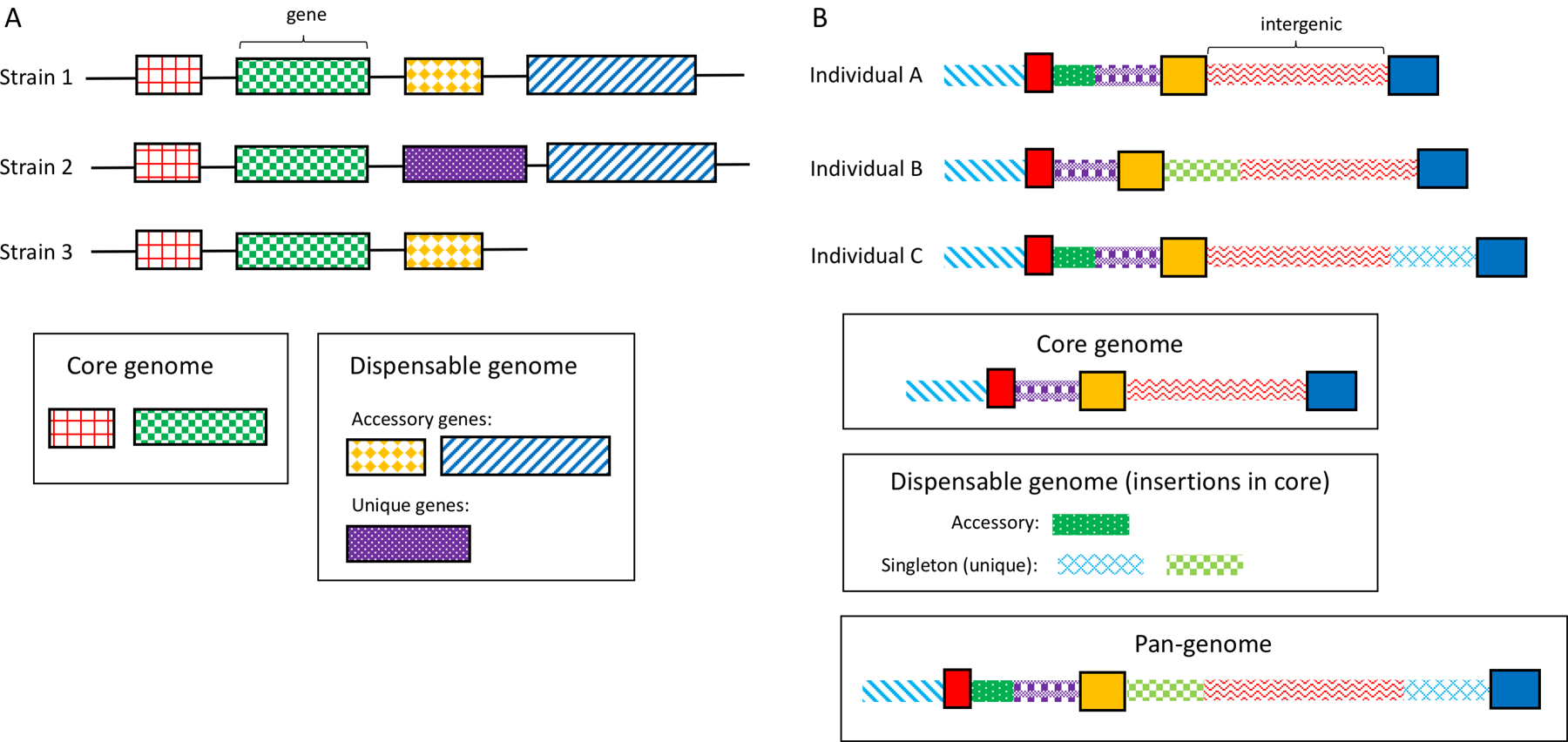
The first work on pan-genomes in bacteria, this paper coined the term ‘pan-genome’ and the associated concepts of the ‘core’ and ‘dispensable’ genomes.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
