

Induced Ketosis as a Treatment for Neuroprogressive Disorders: Food for Thought?
- PMID: 32034911
- PMCID: PMC7311648
- DOI: 10.1093/ijnp/pyaa008
Induced Ketosis as a Treatment for Neuroprogressive Disorders: Food for Thought?
Abstract
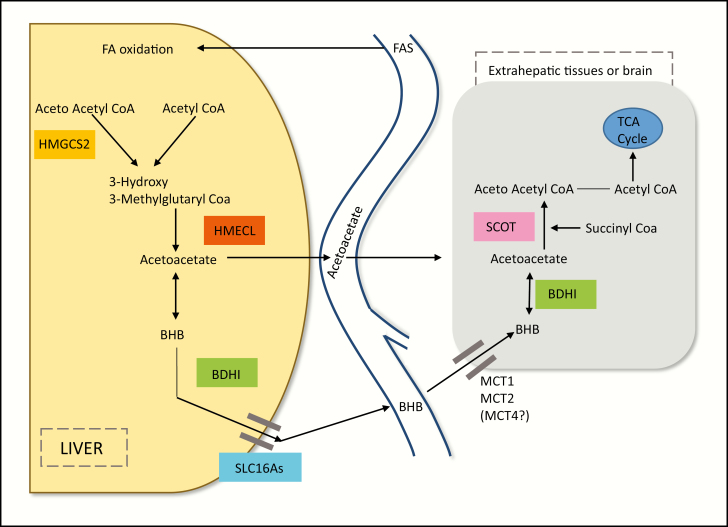
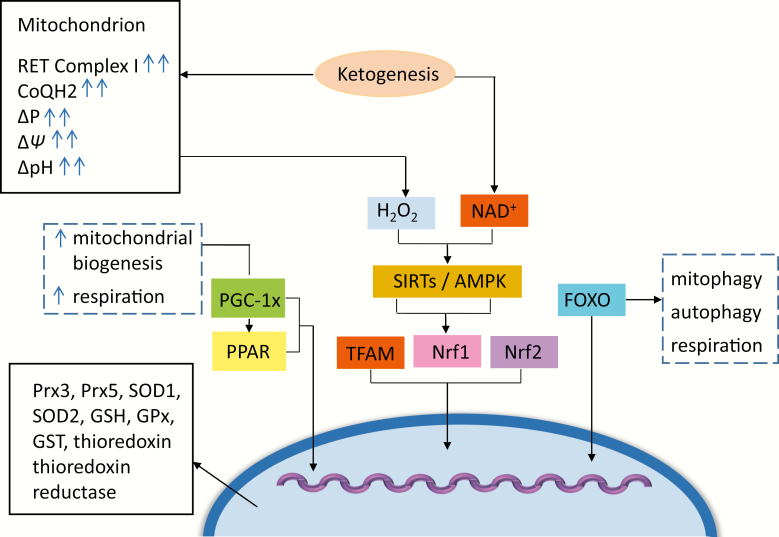
Induced ketosis (or ketone body ingestion) can ameliorate several changes associated with neuroprogressive disorders, including schizophrenia, bipolar disorder, and major depressive disorder. Thus, the effects of glucose hypometabolism can be bypassed through the entry of beta-hydroxybutyrate, providing an alternative source of energy to glucose. The weight of evidence suggests that induced ketosis reduces levels of oxidative stress, mitochondrial dysfunction, and inflammation-core features of the above disorders. There are also data to suggest that induced ketosis may be able to target other molecules and signaling pathways whose levels and/or activity are also known to be abnormal in at least some patients suffering from these illnesses such as peroxisome proliferator-activated receptors, increased activity of the Kelch-like ECH-associated protein/nuclear factor erythroid 2-related factor 2, Sirtuin-1 nuclear factor-κB p65, and nicotinamide adenine dinucleotide (NAD). This review explains the mechanisms by which induced ketosis might reduce mitochondrial dysfunction, inflammation, and oxidative stress in neuropsychiatric disorders and ameliorate abnormal levels of molecules and signaling pathways that also appear to contribute to the pathophysiology of these illnesses. This review also examines safety data relating to induced ketosis over the long term and discusses the design of future studies.
© The Author(s) 2020. Published by Oxford University Press on behalf of CINP.
Figures
Comment in
-
Ketogenic Therapy in Serious Mental Illness: Emerging Evidence.Int J Neuropsychopharmacol. 2020 Jul 29;23(7):434-439. doi: 10.1093/ijnp/pyaa036. Int J Neuropsychopharmacol. 2020. PMID: 32573722 Free PMC article. No abstract available.
Similar articles
-
Nutritional ketosis as an intervention to relieve astrogliosis: Possible therapeutic applications in the treatment of neurodegenerative and neuroprogressive disorders.Eur Psychiatry. 2020 Jan 31;63(1):e8. doi: 10.1192/j.eurpsy.2019.13. Eur Psychiatry. 2020. PMID: 32093791 Free PMC article. Review.
-
The role of microglia in neuroprogressive disorders: mechanisms and possible neurotherapeutic effects of induced ketosis.Prog Neuropsychopharmacol Biol Psychiatry. 2020 Apr 20;99:109858. doi: 10.1016/j.pnpbp.2020.109858. Epub 2020 Jan 7. Prog Neuropsychopharmacol Biol Psychiatry. 2020. PMID: 31923453 Review.
-
Neuroketotherapeutics: A modern review of a century-old therapy.Neurochem Int. 2018 Jul;117:114-125. doi: 10.1016/j.neuint.2017.05.019. Epub 2017 Jun 1. Neurochem Int. 2018. PMID: 28579059 Free PMC article. Review.
-
Medical aspects of ketone body metabolism.Clin Invest Med. 1995 Jun;18(3):193-216. Clin Invest Med. 1995. PMID: 7554586 Review.
-
Impact of Aging on Metabolic Changes in the Ketotic Rat Brain: Glucose, Oxidative and 4-HNE Metabolism.Adv Exp Med Biol. 2018;1072:21-25. doi: 10.1007/978-3-319-91287-5_4. Adv Exp Med Biol. 2018. PMID: 30178318
Cited by
-
Molecular Mechanisms of Neuroprotection by Ketone Bodies and Ketogenic Diet in Cerebral Ischemia and Neurodegenerative Diseases.Int J Mol Sci. 2023 Dec 21;25(1):124. doi: 10.3390/ijms25010124. Int J Mol Sci. 2023. PMID: 38203294 Free PMC article. Review.
-
Role of ketone bodies in diabetes-induced dementia: sirtuins, insulin resistance, synaptic plasticity, mitochondrial dysfunction, and neurotransmitter.Nutr Rev. 2022 Mar 10;80(4):774-785. doi: 10.1093/nutrit/nuab118. Nutr Rev. 2022. PMID: 34957519 Free PMC article. Review.
-
Ketogenic Therapy in Serious Mental Illness: Emerging Evidence.Int J Neuropsychopharmacol. 2020 Jul 29;23(7):434-439. doi: 10.1093/ijnp/pyaa036. Int J Neuropsychopharmacol. 2020. PMID: 32573722 Free PMC article. No abstract available.
-
Brain Metabolism in Health and Neurodegeneration: The Interplay Among Neurons and Astrocytes.Cells. 2024 Oct 17;13(20):1714. doi: 10.3390/cells13201714. Cells. 2024. PMID: 39451233 Free PMC article. Review.
-
The Role of Ketogenic Metabolic Therapy on the Brain in Serious Mental Illness: A Review.J Psychiatr Brain Sci. 2022;7(5):e220009. doi: 10.20900/jpbs.20220009. Epub 2022 Oct 31. J Psychiatr Brain Sci. 2022. PMID: 36483840 Free PMC article.
References
-
- Alageel A, Tomasi J, Tersigni C, Brietzke E, Zuckerman H, Subramaniapillai M, Lee Y, Iacobucci M, Rosenblat JD, Mansur RB, McIntyre RS (2018) Evidence supporting a mechanistic role of sirtuins in mood and metabolic disorders. Prog Neuropsychopharmacol Biol Psychiatry 86:95–101. - PubMed
-
- Bach AC, Ingenbleek Y, Frey A (1996) The usefulness of dietary medium-chain triglycerides in body weight control: fact or fancy? J Lipid Res 37:708–726. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
