

Structural Basis for Lipid Binding and Function by an Evolutionarily Conserved Protein, Serum Amyloid A
- PMID: 32035904
- PMCID: PMC7225066
- DOI: 10.1016/j.jmb.2020.01.029
Structural Basis for Lipid Binding and Function by an Evolutionarily Conserved Protein, Serum Amyloid A
Abstract
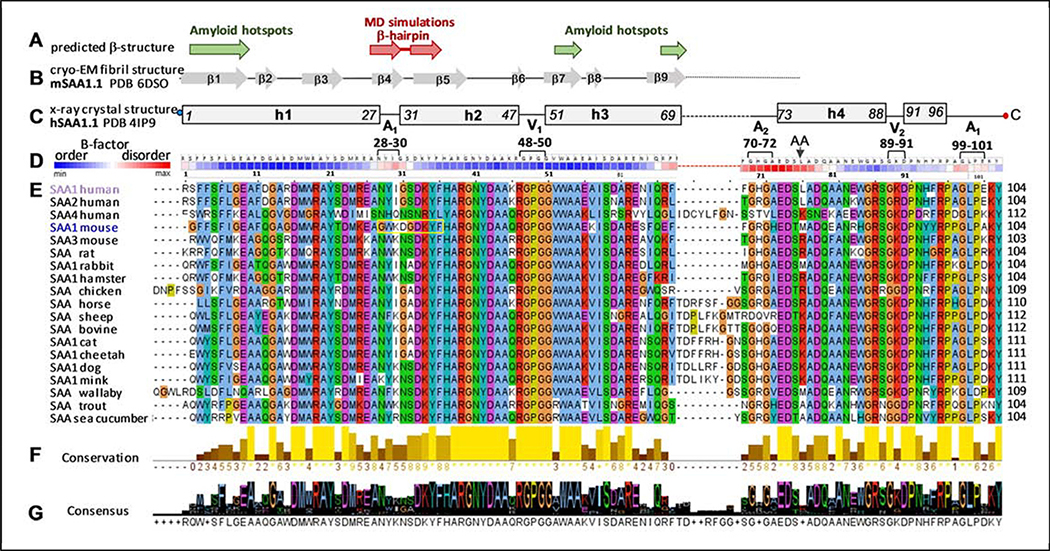
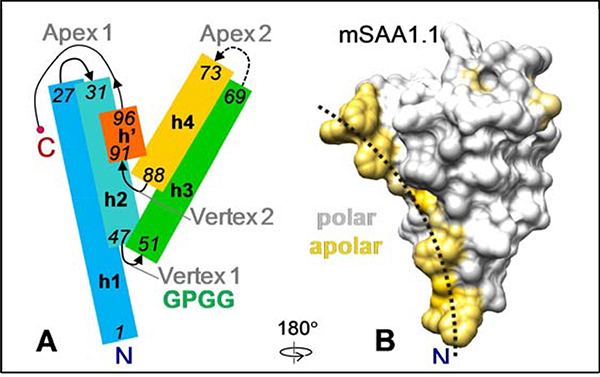
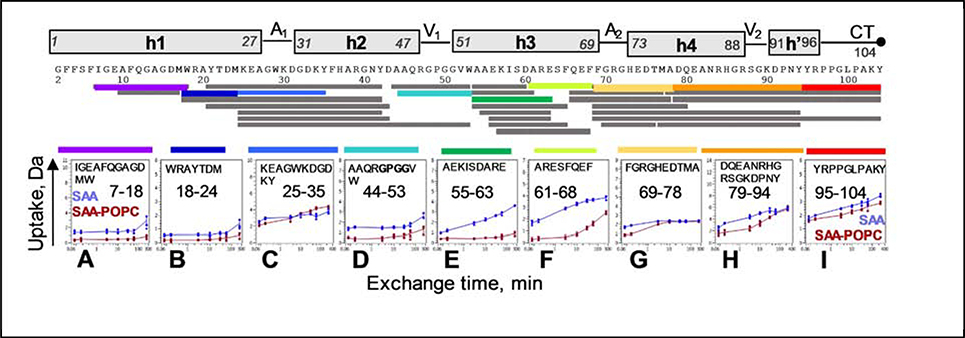
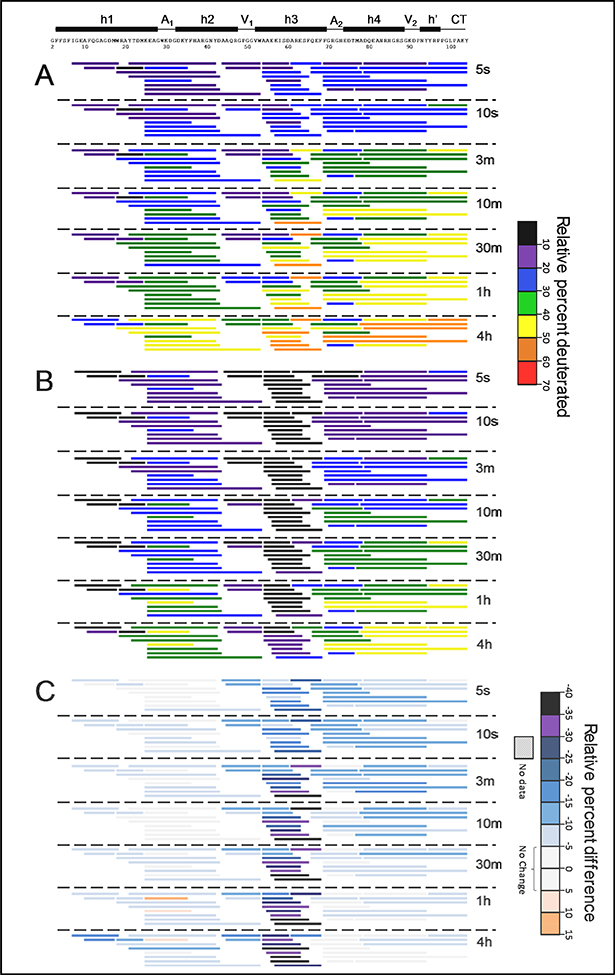
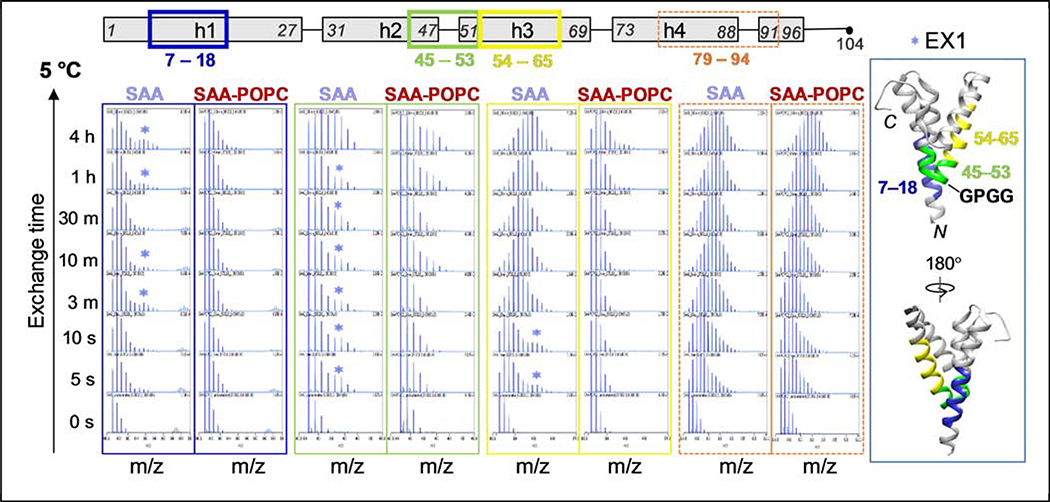
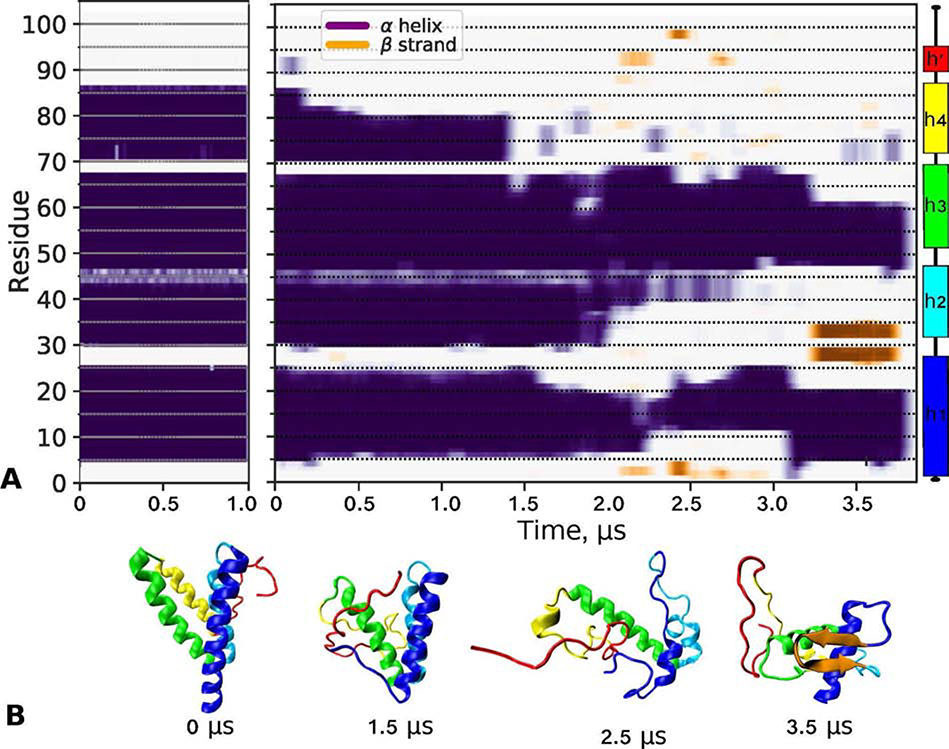
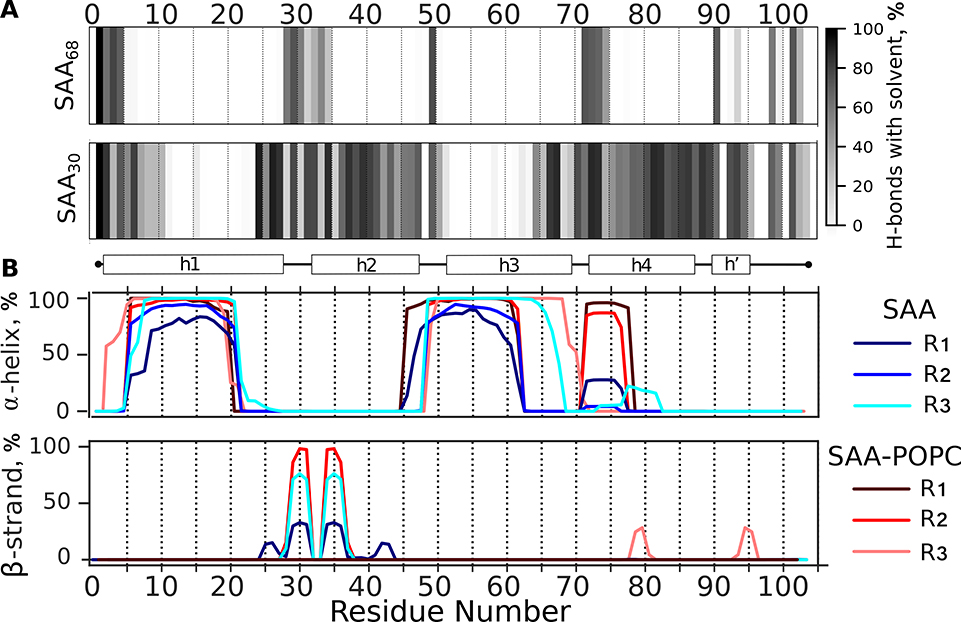
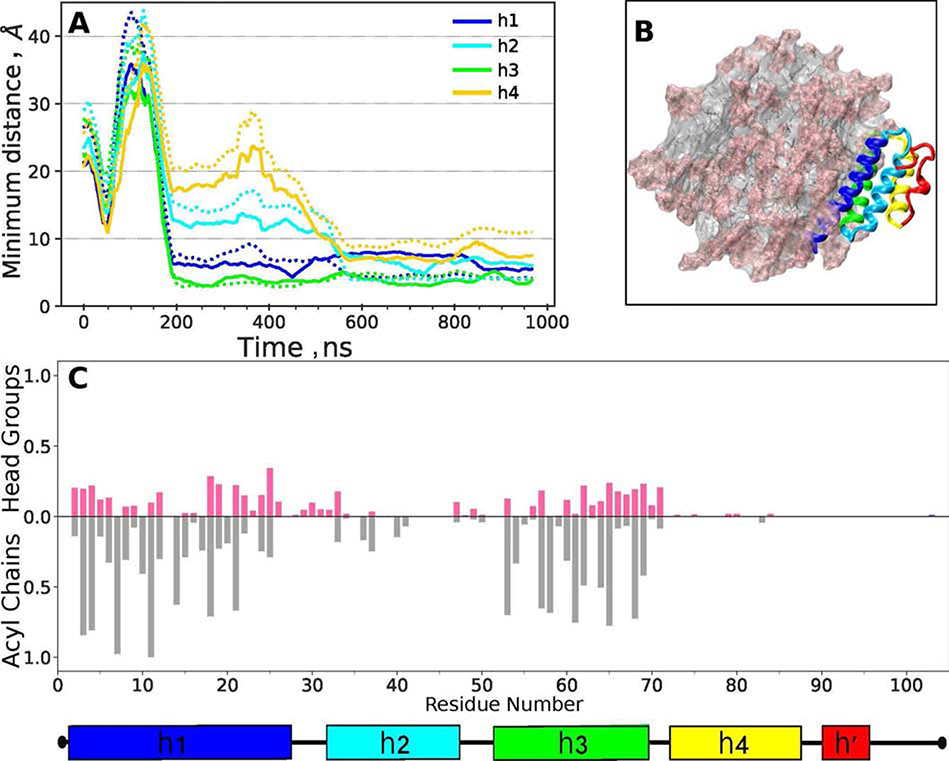
Serum amyloid A (SAA) is a plasma protein that transports lipids during inflammation. To explore SAA solution conformations and lipid-binding mechanism, we used hydrogen-deuterium exchange mass spectrometry, lipoprotein reconstitution, amino acid sequence analysis, and molecular dynamics simulations. Solution conformations of lipid-bound and lipid-free mSAA1 at pH~7.4 agreed in details with the crystal structures but also showed important differences. The results revealed that amphipathic α-helices h1 and h3 comprise a lipid-binding site that is partially pre-formed in solution, is stabilized upon binding lipids, and shows lipid-induced folding of h3. This site sequesters apolar ligands via a concave hydrophobic surface in SAA oligomers. The largely disordered/dynamic C-terminal region is conjectured to mediate the promiscuous binding of other ligands. The h1-h2 linker region is predicted to form an unexpected β-hairpin that may represent an early amyloidogenic intermediate. The results help establish structural underpinnings for understanding SAA interactions with its key functional ligands, its evolutional conservation, and its transition to amyloid.
Keywords: hydrogen-deuterium exchange mass spectrometry; inflammation and immunity; lipoprotein nanoparticle; molecular dynamics simulations; β-hairpin misfolding intermediate.
Copyright © 2020 Elsevier Ltd. All rights reserved.
Figures
References
-
- Urieli-Shoval S, Linke RP, Matzner Y, Expression and function of serum amyloid A, a major acute-phase protein, in normal and disease states, Curr. Opin. Hematol 7 (2000) 64–69. - PubMed
-
- Eklund KK, Niemi K, Kovanen PT, Immune functions of serum amyloid A, Crit. Rev. Immunol 32(4) (2012) 335–348. - PubMed
-
- Westermark GT, Fändrich M, Westermark P, AA amyloidosis: pathogenesis and targeted therapy, Annu. Rev. Pathol 10 (2015) 321–344. - PubMed
-
- Papa R, Lachmann HJ, Secondary AA, Amyloidosis, Rheum. Dis. Clin. North. Am 44(4) (2018) 585–603. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
