

Copy-number variation contributes 9% of pathogenicity in the inherited retinal degenerations
- PMID: 32037395
- PMCID: PMC7272325
- DOI: 10.1038/s41436-020-0759-8
Copy-number variation contributes 9% of pathogenicity in the inherited retinal degenerations
Abstract
Purpose: Current sequencing strategies can genetically solve 55-60% of inherited retinal degeneration (IRD) cases, despite recent progress in sequencing. This can partially be attributed to elusive pathogenic variants (PVs) in known IRD genes, including copy-number variations (CNVs), which have been shown as major contributors to unsolved IRD cases.
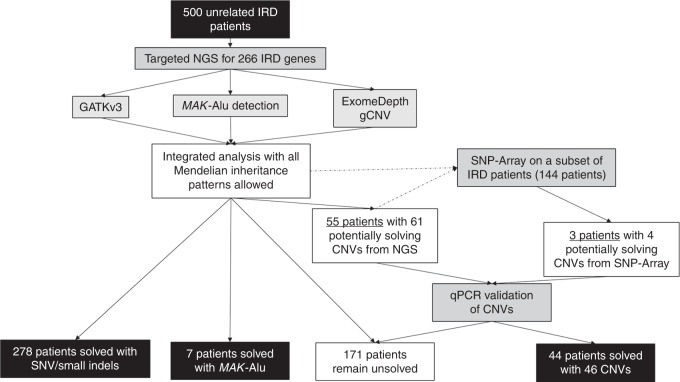
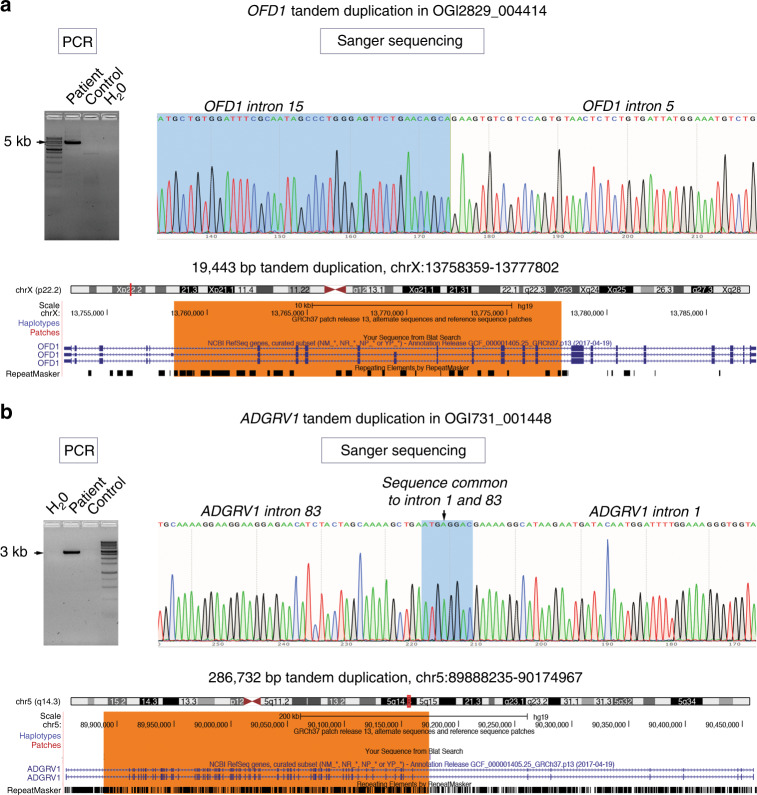
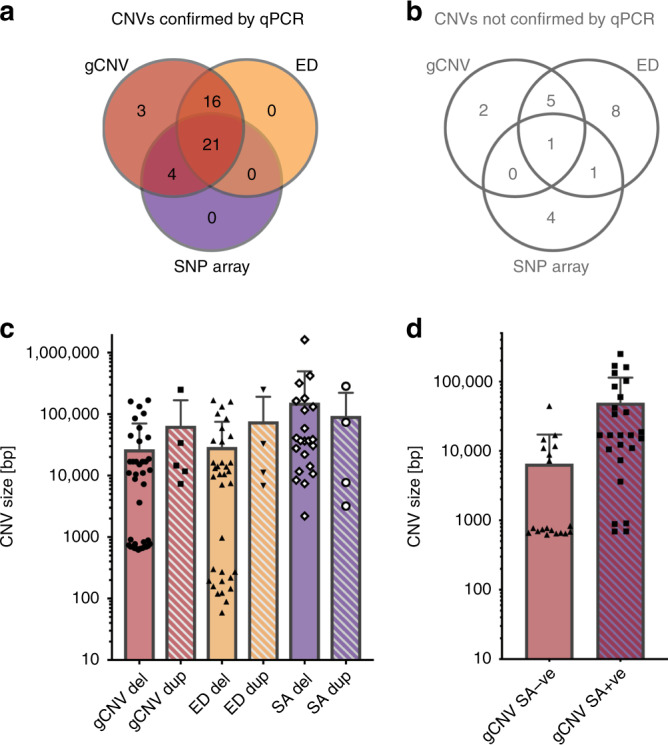
Methods: Five hundred IRD patients were analyzed with targeted next-generation sequencing (NGS). The NGS data were used to detect CNVs with ExomeDepth and gCNV and the results were compared with CNV detection with a single-nucleotide polymorphism (SNP) array. Likely causal CNV predictions were validated by quantitative polymerase chain reaction (qPCR).
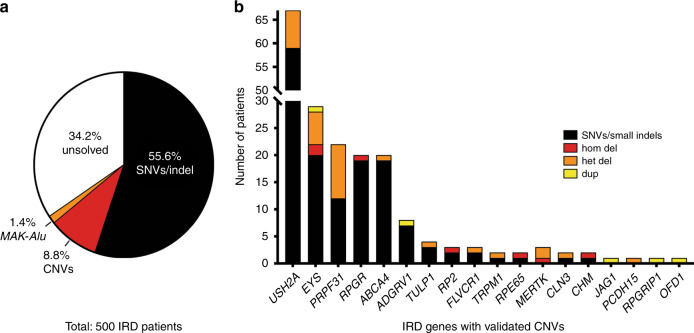
Results: Likely disease-causing single-nucleotide variants (SNVs) and small indels were found in 55.6% of subjects. PVs in USH2A (11.6%), RPGR (4%), and EYS (4%) were the most common. Likely causal CNVs were found in an additional 8.8% of patients. Of the three CNV detection methods, gCNV showed the highest accuracy. Approximately 30% of unsolved subjects had a single likely PV in a recessive IRD gene.
Conclusion: CNV detection using NGS-based algorithms is a reliable method that greatly increases the genetic diagnostic rate of IRDs. Experimentally validating CNVs helps estimate the rate at which IRDs might be solved by a CNV plus a more elusive variant.
Keywords: cone–rod dystrophy; copy-number variation; inherited retinal degeneration; retinitis pigmentosa; rod–cone dystrophy.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
-
- Berger W, Kloeckener-Gruissem B, Neidhardt J. The molecular basis of human retinal and vitreoretinal diseases. Prog Retin Eye Res. 2010;29:335–375. - PubMed
-
- Retinal Information Network. https://sph.uth.edu/retnet/home.htm. 2019. Accessed 20 November 2018.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
