

Role of Endothelial Dysfunction in Cardiovascular Diseases: The Link Between Inflammation and Hydrogen Sulfide
- PMID: 32038245
- PMCID: PMC6985156
- DOI: 10.3389/fphar.2019.01568
Role of Endothelial Dysfunction in Cardiovascular Diseases: The Link Between Inflammation and Hydrogen Sulfide
Abstract
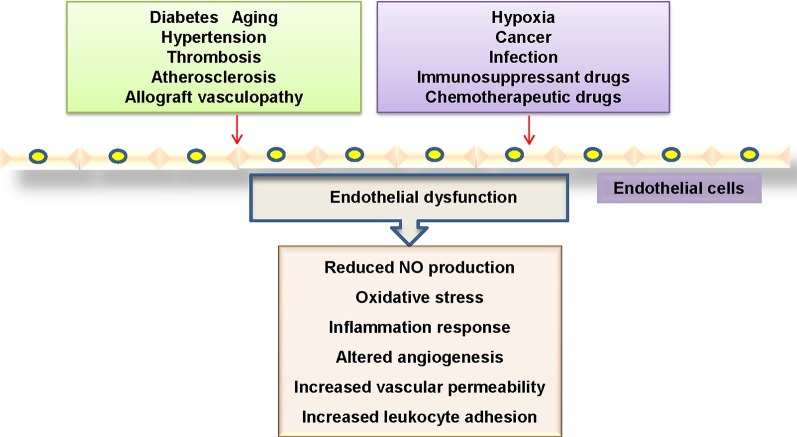
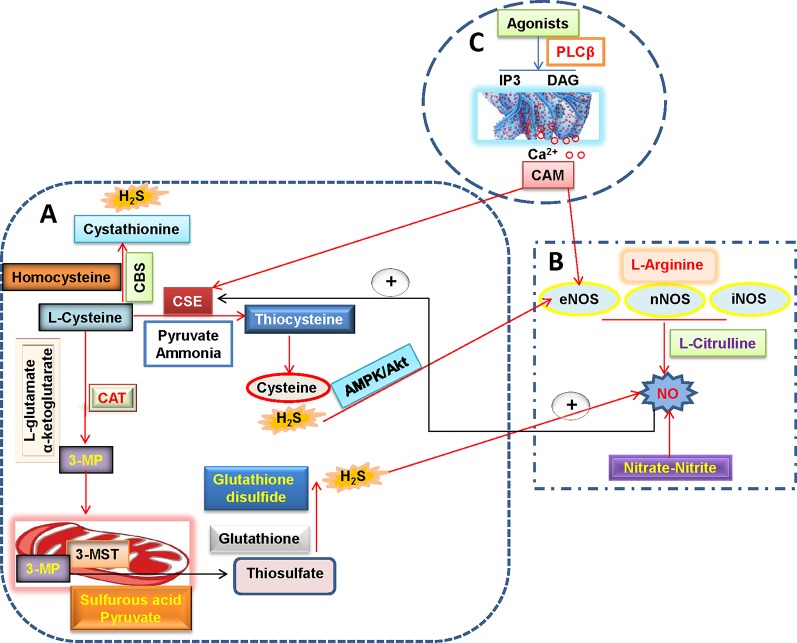
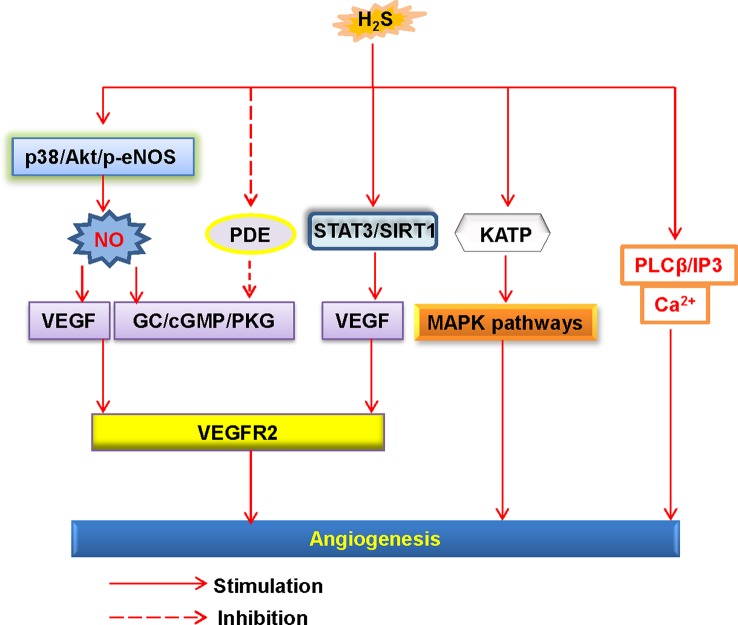
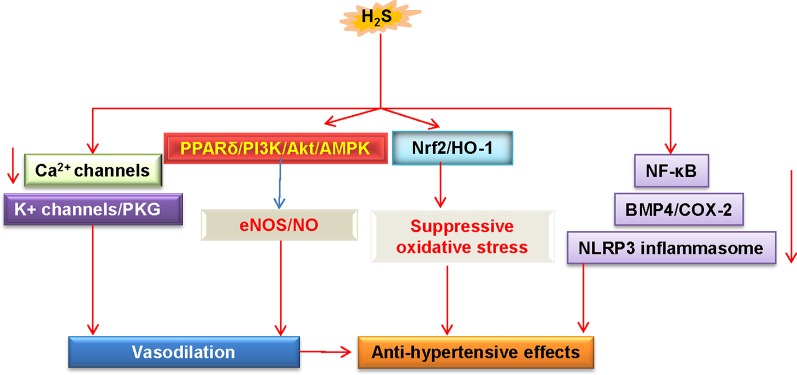
Endothelial cells are important constituents of blood vessels that play critical roles in cardiovascular homeostasis by regulating blood fluidity and fibrinolysis, vascular tone, angiogenesis, monocyte/leukocyte adhesion, and platelet aggregation. The normal vascular endothelium is taken as a gatekeeper of cardiovascular health, whereas abnormality of vascular endothelium is a major contributor to a plethora of cardiovascular ailments, such as atherosclerosis, aging, hypertension, obesity, and diabetes. Endothelial dysfunction is characterized by imbalanced vasodilation and vasoconstriction, elevated reactive oxygen species (ROS), and proinflammatory factors, as well as deficiency of nitric oxide (NO) bioavailability. The occurrence of endothelial dysfunction disrupts the endothelial barrier permeability that is a part of inflammatory response in the development of cardiovascular diseases. As such, abrogation of endothelial cell activation/inflammation is of clinical relevance. Recently, hydrogen sulfide (H2S), an entry as a gasotransmitter, exerts diverse biological effects through acting on various targeted signaling pathways. Within the cardiovascular system, the formation of H2S is detected in smooth muscle cells, vascular endothelial cells, and cardiomyocytes. Disrupted H2S bioavailability is postulated to be a new indicator for endothelial cell inflammation and its associated endothelial dysfunction. In this review, we will summarize recent advances about the roles of H2S in endothelial cell homeostasis, especially under pathological conditions, and discuss its putative therapeutic applications in endothelial inflammation-associated cardiovascular disorders.
Keywords: cardiovascular disease; endothelial cell; gasotransmitters; hydrogen sulfide; inflammation.
Copyright © 2020 Sun, Wu, Nie and Bian.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Research Materials