

De novo protein design, a retrospective
- PMID: 32041676
- PMCID: PMC7243446
- DOI: 10.1017/S0033583519000131
De novo protein design, a retrospective
Abstract
Proteins are molecular machines whose function depends on their ability to achieve complex folds with precisely defined structural and dynamic properties. The rational design of proteins from first-principles, or de novo, was once considered to be impossible, but today proteins with a variety of folds and functions have been realized. We review the evolution of the field from its earliest days, placing particular emphasis on how this endeavor has illuminated our understanding of the principles underlying the folding and function of natural proteins, and is informing the design of macromolecules with unprecedented structures and properties. An initial set of milestones in de novo protein design focused on the construction of sequences that folded in water and membranes to adopt folded conformations. The first proteins were designed from first-principles using very simple physical models. As computers became more powerful, the use of the rotamer approximation allowed one to discover amino acid sequences that stabilize the desired fold. As the crystallographic database of protein structures expanded in subsequent years, it became possible to construct proteins by assembling short backbone fragments that frequently recur in Nature. The second set of milestones in de novo design involves the discovery of complex functions. Proteins have been designed to bind a variety of metals, porphyrins, and other cofactors. The design of proteins that catalyze hydrolysis and oxygen-dependent reactions has progressed significantly. However, de novo design of catalysts for energetically demanding reactions, or even proteins that bind with high affinity and specificity to highly functionalized complex polar molecules remains an importnant challenge that is now being achieved. Finally, the protein design contributed significantly to our understanding of membrane protein folding and transport of ions across membranes. The area of membrane protein design, or more generally of biomimetic polymers that function in mixed or non-aqueous environments, is now becoming increasingly possible.
Keywords: Protein design.
Figures
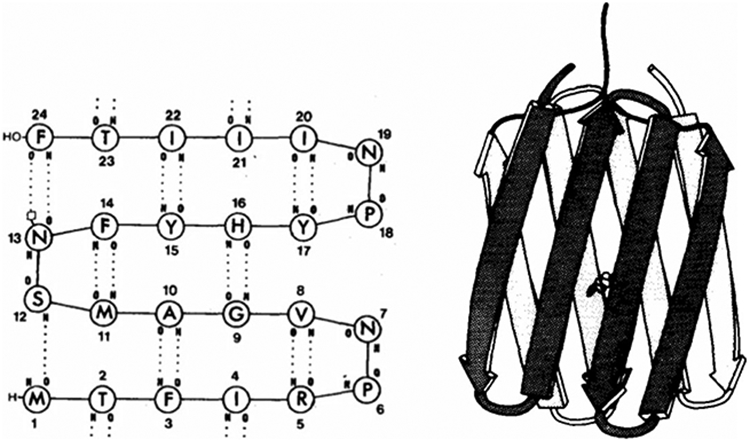
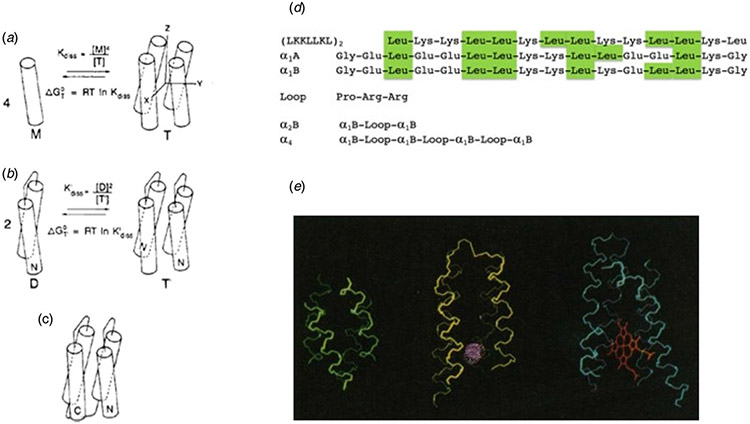
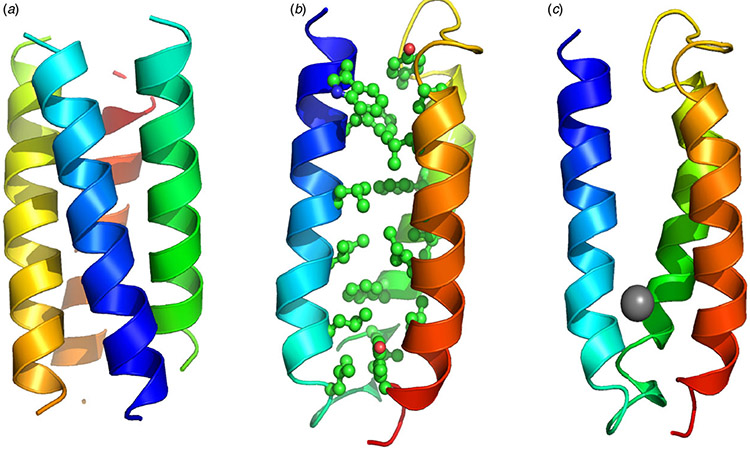
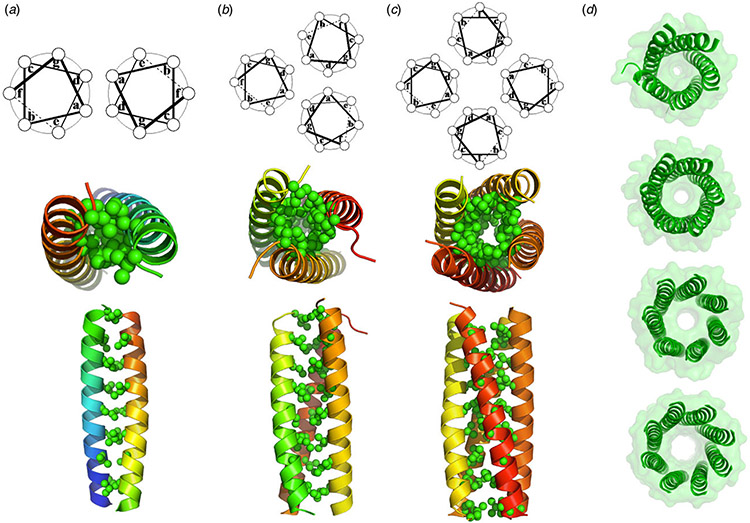
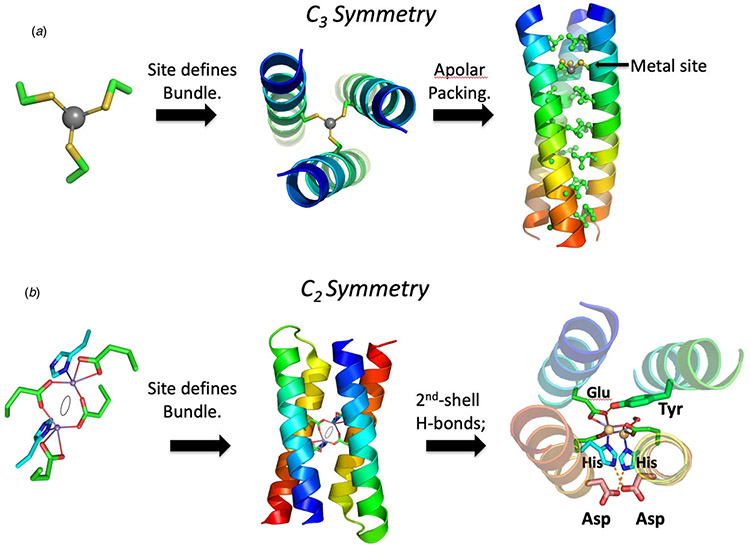
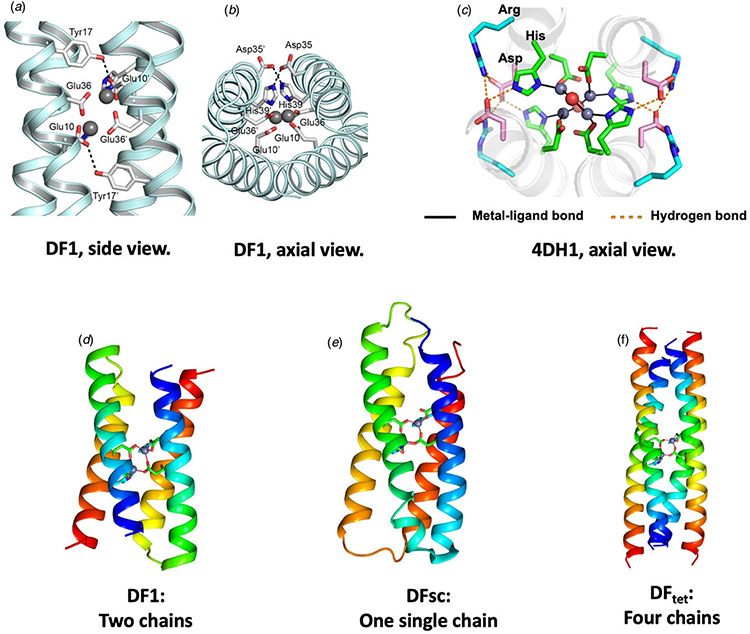
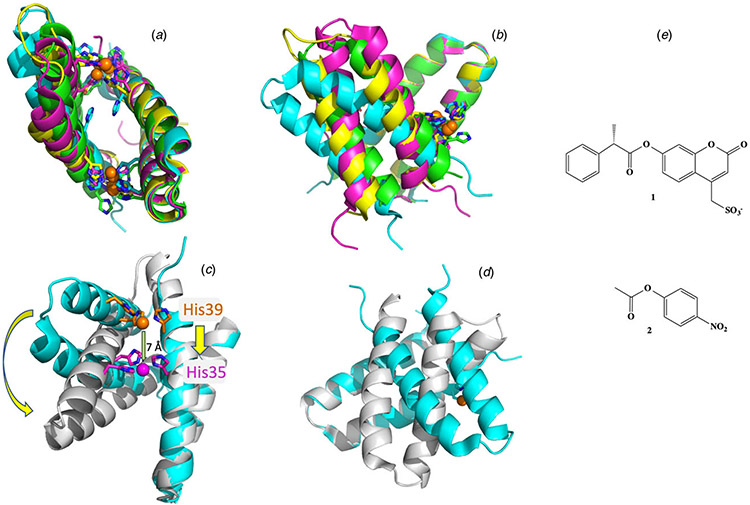
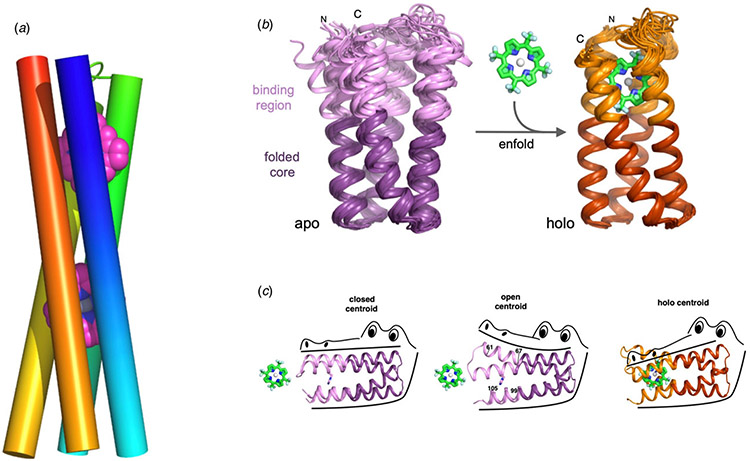
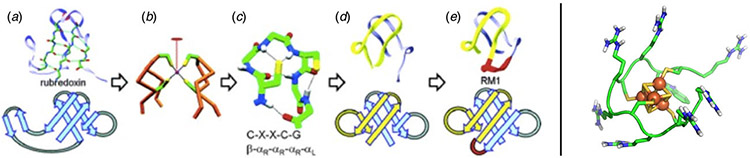
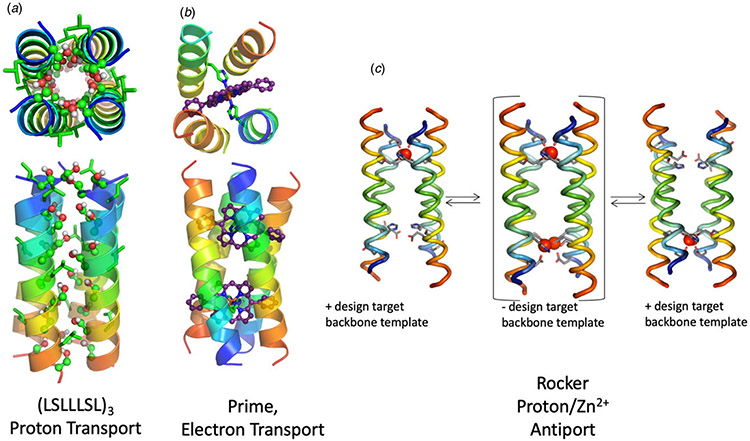
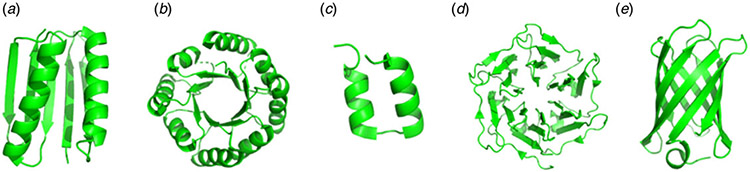
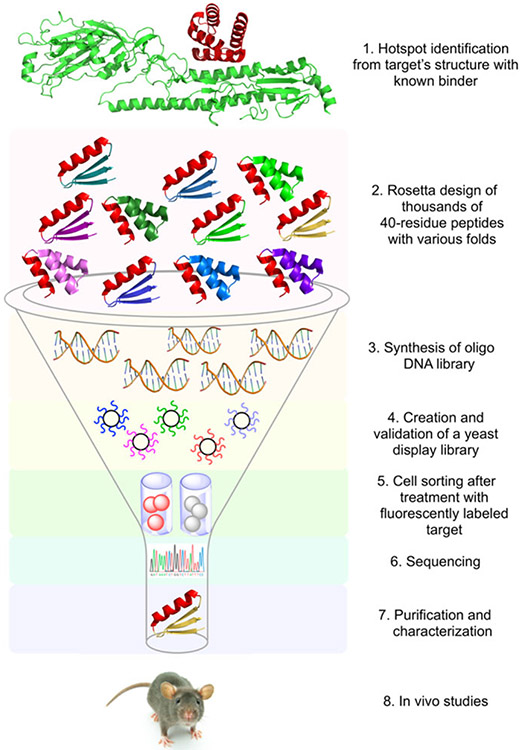
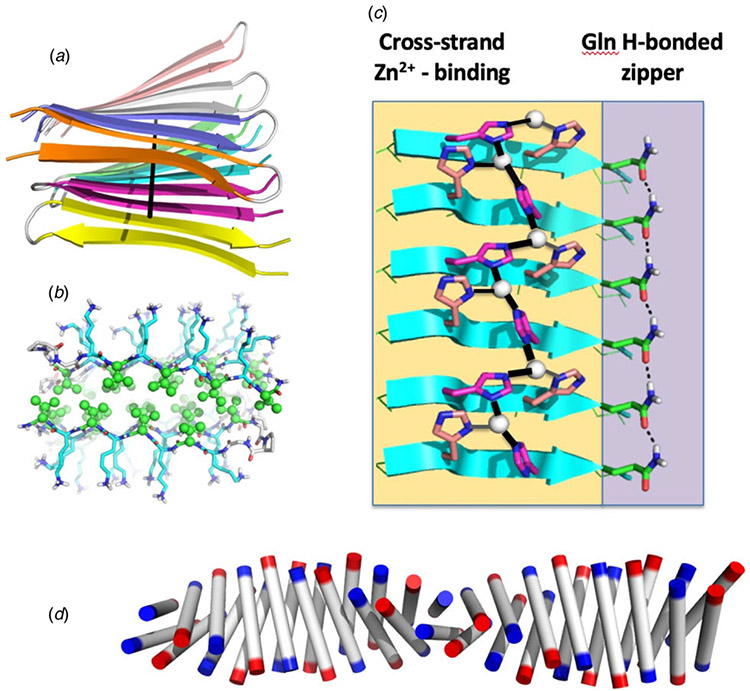
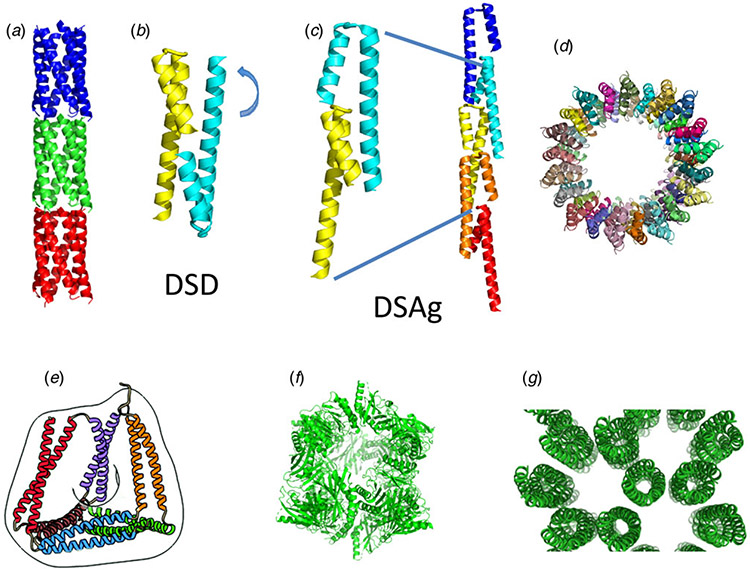
References
-
- Adamian L and Liang J (2001) Helix–helix packing and interfacial pairwise interactions of residues in membrane proteins. Journal of Molecular Biology 311, 891–907. - PubMed
-
- Adamian L and Liang J (2002) Interhelical hydrogen bonds and spatial motifs in membrane proteins: polar clamps and serine zippers. Proteins 47, 209–218. - PubMed
-
- Åkerfeldt K, Kim RM, Camac D, Groves JT, Lear JD and DeGrado WF (1992) Tetraphilin: a four-helix proton channel built on a tetraphenylporphyrin framework. Journal of the American Chemical Society 114, 9656–9657.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
