

Genome-wide meta-analysis identifies eight new susceptibility loci for cutaneous squamous cell carcinoma
- PMID: 32041948
- PMCID: PMC7010741
- DOI: 10.1038/s41467-020-14594-5
Genome-wide meta-analysis identifies eight new susceptibility loci for cutaneous squamous cell carcinoma
Abstract
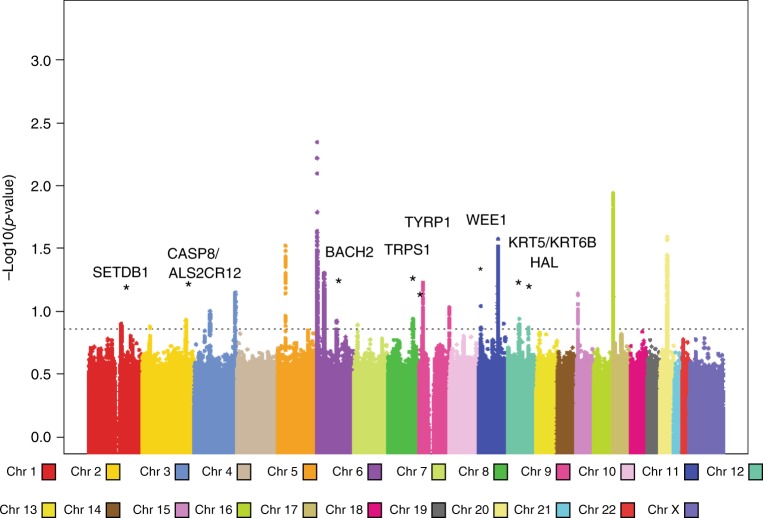
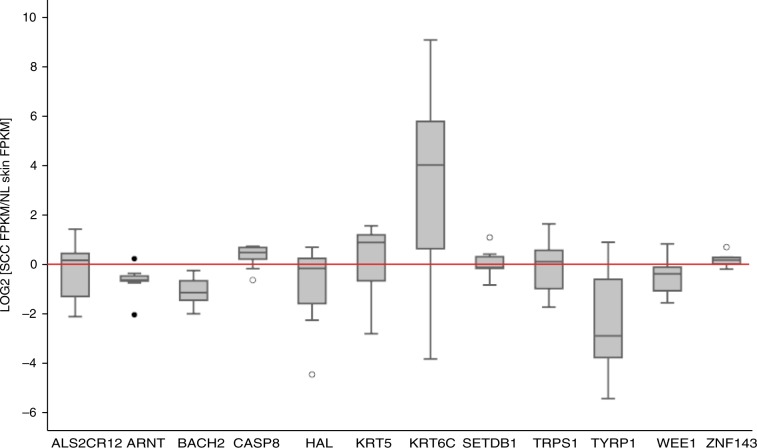
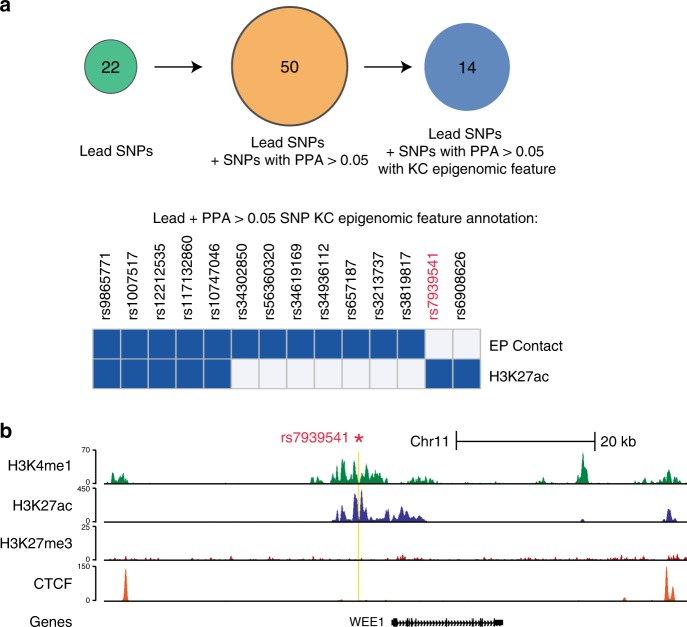
Cutaneous squamous cell carcinoma (SCC) is one of the most common cancers in the United States. Previous genome-wide association studies (GWAS) have identified 14 single nucleotide polymorphisms (SNPs) associated with cutaneous SCC. Here, we report the largest cutaneous SCC meta-analysis to date, representing six international cohorts and totaling 19,149 SCC cases and 680,049 controls. We discover eight novel loci associated with SCC, confirm all previously associated loci, and perform fine mapping of causal variants. The novel SNPs occur within skin-specific regulatory elements and implicate loci involved in cancer development, immune regulation, and keratinocyte differentiation in SCC susceptibility.
Conflict of interest statement
G.T., S.N.S. and K.S. are employees of deCODE Genetics.
Figures
Similar articles
-
Combined analysis of keratinocyte cancers identifies novel genome-wide loci.Hum Mol Genet. 2019 Sep 15;28(18):3148-3160. doi: 10.1093/hmg/ddz121. Hum Mol Genet. 2019. PMID: 31174203 Free PMC article.
-
Multi-ancestry genome-wide meta-analysis identifies novel basal cell carcinoma loci and shared genetic effects with squamous cell carcinoma.Commun Biol. 2024 Jan 5;7(1):33. doi: 10.1038/s42003-023-05753-7. Commun Biol. 2024. PMID: 38182794 Free PMC article.
-
Genome-wide association study identifies novel susceptibility loci for cutaneous squamous cell carcinoma.Nat Commun. 2016 Jul 18;7:12048. doi: 10.1038/ncomms12048. Nat Commun. 2016. PMID: 27424798 Free PMC article.
-
Genome-wide association studies of pigmentation and skin cancer: a review and meta-analysis.Pigment Cell Melanoma Res. 2010 Oct;23(5):587-606. doi: 10.1111/j.1755-148X.2010.00730.x. Epub 2010 Jul 16. Pigment Cell Melanoma Res. 2010. PMID: 20546537 Free PMC article. Review.
-
Lessons from postgenome-wide association studies: functional analysis of cancer predisposition loci.J Intern Med. 2013 Nov;274(5):414-24. doi: 10.1111/joim.12085. J Intern Med. 2013. PMID: 24127939 Free PMC article. Review.
Cited by
-
The Immunogenetics of Non-melanoma Skin Cancer.Adv Exp Med Biol. 2022;1367:397-409. doi: 10.1007/978-3-030-92616-8_16. Adv Exp Med Biol. 2022. PMID: 35286705
-
Polygenic risk scores, radiation treatment exposures and subsequent cancer risk in childhood cancer survivors.Nat Med. 2024 Mar;30(3):690-698. doi: 10.1038/s41591-024-02837-7. Epub 2024 Mar 7. Nat Med. 2024. PMID: 38454124 Free PMC article.
-
Polygenic Risk and Chemotherapy-Related Subsequent Malignancies in Childhood Cancer Survivors: A Childhood Cancer Survivor Study and St Jude Lifetime Cohort Study Report.J Clin Oncol. 2023 Sep 20;41(27):4381-4393. doi: 10.1200/JCO.23.00428. Epub 2023 Jul 17. J Clin Oncol. 2023. PMID: 37459583 Free PMC article.
-
A custom capture sequence approach for oculocutaneous albinism identifies structural variant alleles at the OCA2 locus.Hum Mutat. 2021 Oct;42(10):1239-1253. doi: 10.1002/humu.24257. Epub 2021 Aug 1. Hum Mutat. 2021. PMID: 34246199 Free PMC article.
-
Assessment of the Influence of UVR in Cutaneous Melanoma.Photodermatol Photoimmunol Photomed. 2025 May;41(3):e70024. doi: 10.1111/phpp.70024. Photodermatol Photoimmunol Photomed. 2025. PMID: 40396496 Free PMC article. Review.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
